 With the clock for the countdown to the end of transitional periods for the MDR and IVDR ticking away, everyone is of course very interested what the competent authorities are doing regarding implementation of the MDR and IVDR, since a lot needs to happen to implement the MDR and IVDR. The Dutch competent authority gave some insight in planning earlier this year at a seminar organised by my firm.
With the clock for the countdown to the end of transitional periods for the MDR and IVDR ticking away, everyone is of course very interested what the competent authorities are doing regarding implementation of the MDR and IVDR, since a lot needs to happen to implement the MDR and IVDR. The Dutch competent authority gave some insight in planning earlier this year at a seminar organised by my firm.
The work to be done concerns three main areas:
- delegated and implementing acts defined in the MDR and IVDR to make the system work (such as regarding the functioning of Eudamed and UDI);
- EU level guidance on the many new concepts and procedures; and
- National implementation in the fields where there is national discretion in implementation (e.g. in the field of whether and how to permit reprocessing).
The competent authorities have been busy in the background and have now produced a roadmap for implementation, defining priorities for seven technical areas and some horizontal / cross cutting issues.
Wait what? With 1/6th of the MDR transitional period already passed they are only starting to define priorities for implementation now? With not everything high priority or with even a fixed date attached? Yes, indeed and unfortunately – so better keep a close watch on the development and be ready to act quickly when the documents become available.
The document gives a good overview of responsible parties for each of the items in the lists, allowing you to see the new governance structure under the MDCG at work. 26 N November is also a memorable date (not only because it’s the date on which notified bodies can apply for MDR accreditation) because it’s the date as of which the MDCG is formally operational (see article 103 jo 123 (1) (b) MDR) and can start to get stuff formally organised for the big rollout program for the MDR and IVDR. Yay! We will then soon know more about its members and it will allow the newly set up MDCG to take a number of actions that are currently brewing in the background, which can be any of the things the MDCG is allowed to do, see articles 103 and 105 MDR / 99 IVDR.
The seven priority areas are:
- Clinical Evaluation & Clinical Investigation (MD); Performance Evaluation & Performance Studies (IVD)
- Scope & Classification
- Notified Bodies & Conformity Assessment
- Post-Market Surveillance & Vigilance for both MD and IVD
- Eudamed & UDI
- Market Surveillance
- IVD-specific Issues
Let’s take a look each of these, and then discuss the horizontal priorities at the end.
Clinical Evaluation & Clinical Investigation (MD); Performance Evaluation & Performance Studies (IVD)
The MDR is about More Data Really (and the IVDR too) – that data being specifically clinical (MDR) and clinical performance (IVDR) data. It’s not surprising therefore that an important part of the roadmap is concerned with clinical and performance data and how to deal with it for the purpose of evaluation, conformity assessment and post-market follow-up.
This section of the document is part is the product of the Clinical Investigation and Evaluation Working Group. The plan is to develop templates for several clinical related deliverables prescribed in the MDR and IVDR, and its high priority:
- Summary of Safety and Clinical Performance (MD)
- Summary of Safety and Performance (IVD)
- Clinical Evaluation Assessment Report (MD)
- Performance evaluation plan and performance evaluation report (IVD)
- Clinical Investigation application form (MD)
- CI Assessment Report (MD)
- Performance study Application Form (IVD)
- Performance Study Report (IVD)
- SAE/device deficiency reports and timelines (MD and IVD)
- PMCF plan and PMCF report (MD)
- PMPF plan and PMPF report (IVD)
I have understood that the recent 6 November meeting of the working group confirmed that MEDDEV 2.1/1 rev 4 really is guidance and not to be religiously applied written in stone rules (basically endorsing the view TUV SUD took earlier this year).
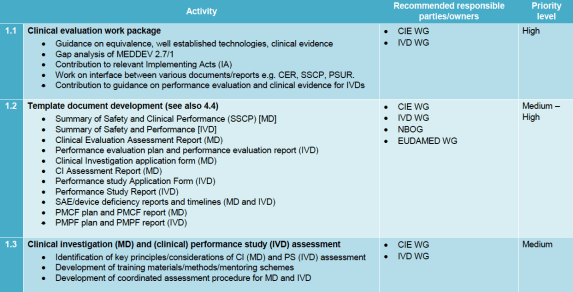
The working group is working on a procedure for developing ‘device specific guidance’, which we should see as the precursor of new common specifications that can be issued under the MDR / IVDR. A draft of device specific guidance for drug eluting stents and bioresorbable stents is in the works. It’s not surprising that they started with stents, because some EU consensus on clinical evaluation of coronary stents already exists. Developing common specifications is also one of the actions under the roadmap’s clinical chapters.
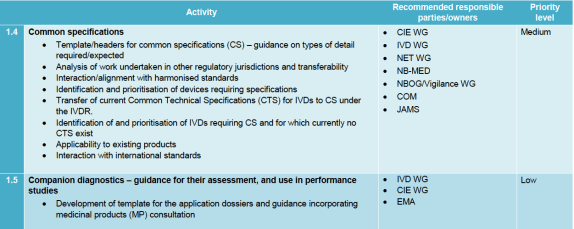
Another item in the works is the development of template for the application dossiers and guidance incorporating medicinal products (MP) consultation, but this has low priority.
I hope this is the start of more harmonisation of clinical evaluation in the EU. We will see this come from the clinical evaluation consultation procedure (the procedure formerly known as scrutiny) and the development of common specifications, but any other harmonisation is welcome too, as it makes the CE marking process more predictable.
For more details, see the entire table in the roadmap document.
Scope & Classification
The items for scope and classification are as follows:
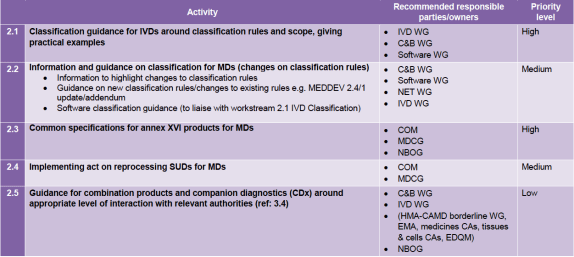
An interesting point about the combination products and guidance is the issue that the no grandfathering principle raised for MDR certification of existing combination products. Does that mean that the full procedure (210 days) for medicine consultation has to be followed as referred to in Annex IX, 5.2 sub (d) for conformity assessment of devices incorporating a medicinal substance, or is this a ‘change’ that takes 60 days under Annex IX, 5.2 sub (f)? Nobody is sure yet for lack of an official position.
Another interesting one is that there seems to be a developing consensus that the combination products procedure under Annex IX, 5.2 also applies to rule 21 substance based devices.
Notified Bodies & Conformity Assessment
By now it becomes more and more clear that a lot of the initial assumptions about what notified bodies were going to apply when for what regulation and what scope are very fluid with the 26 November application opening date around the corner. Traditional notified bodies are applying later or not, or in a limited scope and new notified bodies enter the scene, with even some consultancies and companies applying for targeted scopes.
What is standard is that the notified bodies currently in the market are routinely vague to the point of being misleading to their (potential) clients about what they are actually going to do, and no notified body is very happy to tell you the exact scope for which they are applying. This makes is basically impossible to compare notified bodies, which is one of the big points in the legislative history of the MDR and IVDR with respect to the notified bodies: they do not actually compete because prices are not transparent, but now there is this too. So I decided to file a freedom of information request with the Commission after 26 November to ask the Commission to disclose information on this point so we can all benefit from some transparency. Keep a watch on this blog for the results.

Not surprisingly many of the notified body related actions are high priority, like guidance to be issued on designation process for joint assessments under the new regulations. Yes, that would be nice – to have some guidance on this critical process. Better already too late than never right? The process of designation will be complex enough. Rather than create synergies the assessment under the MDR, the IVDR and the joint assessment for the current directives will happen each separately, putting an enormous strain on the limited competent authority resources.
An interesting one is the guidance on “What is a significant change?”. This will be crucial if you are going to rely on the ‘soft transition’ period, under which you can rely on a certificate under the old directives running past the date of application, provided that there is no ‘significant change’ to the device design or intended purpose. This is also an item below under the horizontal subjects. We know that there is some controversy between the member states on the exact interpretation of the concept of significant change, which may delay the formation of the guidance on this concept.
Guidance on procedure for notified bodies is also welcome, as it may provide much needed streamlining of conformity assessment for MDR and IVDR certificates towards the end of the transitional periods.
Post-Market Surveillance & Vigilance for both MD and IVD
In relation to post-market surveillance an important one is updated vigilance guidance (this will change compared to the current directives) and related terminology (you’ll need guidance indeed if you change so many well-established concepts – duh).
New template forms to replace the ones currently attached to MEDDEV 2.12-1 Rev 8 on vigilance will be developed too.

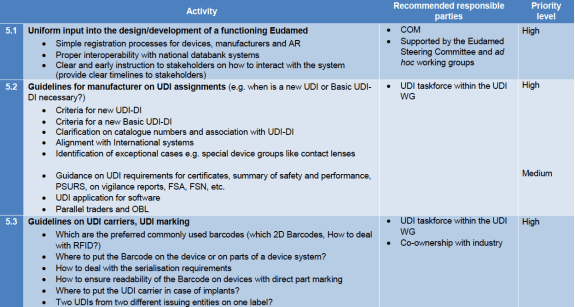
Eudamed & UDI
In relation to Eudamed the million Euro question is of course if Eudamed will be ready in time and actually work. The focus in the roadmap is on interaction with Eudamed and UDI operational matters. Don’t forget though that even if Eudamed is not ready, the only obligation that the manufacturers will not have is actually inputting information in Eudamed. All the other obligations, like obtaining SRNs (you need one for application for conformity assessment for example, see article 31 (3) MDR) and also assigning UDI to devices, because that obligation is independent of registering the device in the database under its UDI.
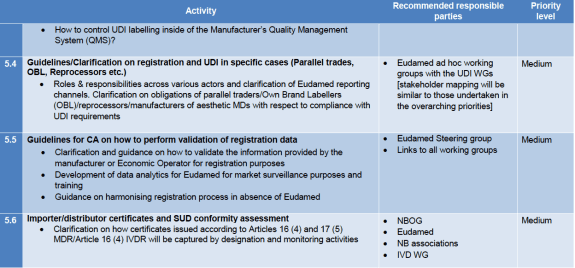
Market Surveillance
Nothing is urgent in market surveillance – what a relief. That doesn’t mean that not a lot is going to happen as the member states build up and tie together the whole EU enforcement backoffice. This is not a direct implementation issue for companies, but it is something to keep track of, for example to understand how the authorities will use clinical information as part of market surveillance, and how they will review clinical evaluation, performance evaluation and the post market data collection processes (last line under 6.5 below).
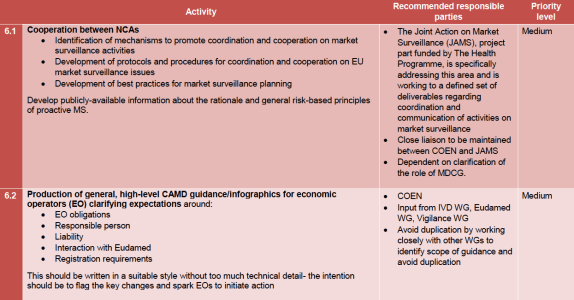
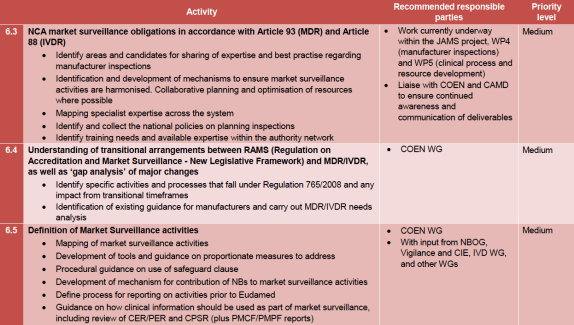
IVD-specific Issues
Most of the IVD specific issues also feature in the other streams above as you can see in the table. The only urgent priority are the reference laboratories – their designation and their function (see also below under 8.7 at the horizontal issues). Indeed – they are a gating issue for all class D IVDs for which there are no common specifications yet (IVD ‘scrutiny’ procedure) and there will need to be a common standard for assessing those, which there currently is not. The reference labs also play an important role under the IVDR to verify by by laboratory testing for conformity testing purposes the performance claimed by the manufacturer and the compliance of devices presenting the highest risk with the applicable CS (when available), or with other solutions chosen by the manufacturer to ensure a level of safety and performance that is at least equivalent.
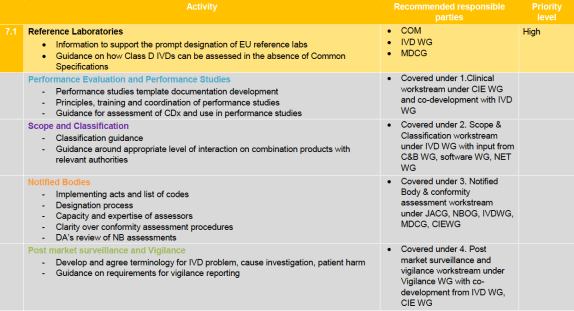

It’s good to see an action item for a template for performance studies documentation and how to prepare and perform them. Given that most of the IVDs currently self-certified will need to go through notified body assessment based on significantly more and better presented data than currently likely available, this guidance will be very useful. Also the classification guidance will be useful because the IVD sector will need to learn to work with the new classification rules under the IVDR.
Notified body expertise and capacity for IVDs under the IVDR will be a very big item as I have observed before, so to me it comes at no surprise that this is an item for the CAMD roadmap too.
Over-arching & Cross-cutting Priorities
While sections 1 to 7 of the roadmap were sort of known already since April the section 8 on the over-arching and cross-cutting priorities is new. They are indeed priorities because everything in this section is high priority or even – exceptionally – has a date assigned to it (that’s Eudamed being ready).
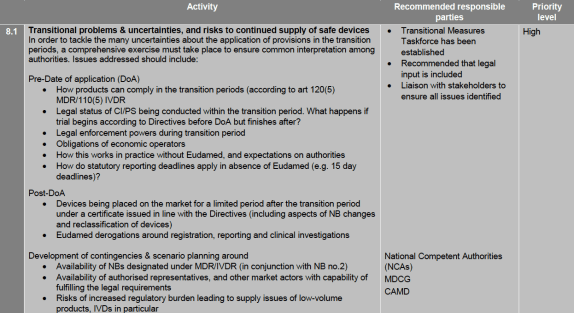
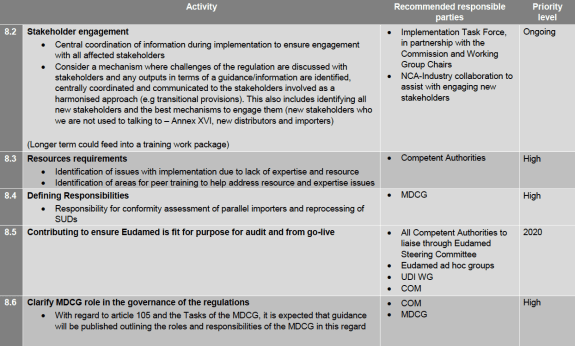

Interesting elements are the pre-date of application (DoA) and post-DoA questions, most of which have been raised on this blog already one way or another (I hope you’ve been paying attention). It will be good that there is guidance that companies can follow though in order to manage contingencies.
Contingency scenario development and planning is another interesting one, especially the supply problems that can take place in cases of low volume products. I would add to these the case of crucial healthcare software that is catapulted from class I to class III and does not manage to get certified in time and cannot benefit from the soft transition period. As things are in the software market single software suites can become a standard by themselves. If that software suddenly needs to be taken off the market as corrective measure (e.g. because of a critical patch that needs to be implement but cannot be because it changes the software installed base) or cannot be made available after the DoA this can have serious implications for health institutions.
How for do?
Please do not make the mistake to think that the fact that all of the above rollout and implementation roadmap is in development means that you can sit on your hands and wait until the documents have been released. You are smarter than an ostrich, right? A smart medical devices company is already doing what it can to be ready in time and sees all the guidance that will come out at some point as a welcome benchmarking exercise. If you start implementing by the time the guidance may be out (and we don’t know when that is), you will likely be too late to meaningfully act on it.
In way companies can see this roadmap as an overview of questions that they should be asking themselves in the frame of their transition program towards MDR and/or IVDR compliance. The specifics where guidance is developed are specifics that the company must pay extra attention to itself.
Companies that are underway with their transition process can take this in stride and supplement what they are doing already with the new guidance as and when it becomes available. Better pivot a little halfway than start too late to be in time, my late grandmother would say.
Workshop

In any event, if you’re based in the US, you could consider visiting the EU MDR and IVDR transition workshop that I’m giving at Advamed in Washington DC on 4 December to get the full picture and be prepared.
I will go into this stuff in a lot more detail in person too and you can ask any questions you have! Regardless of where you are in your transitioning process this workshop will be worth your while.



3 comments
Do you want to comment?
Comments RSS and TrackBack URI
Trackbacks