
You must be thinking that this blog is a bit quiet while a lot is happing – and that’s exactly the case. I’ve been very very busy being Batman (see picture on the left).
A lot of companies in devices still do not seem to get what the enormous change brought about by the MDR and IVDR will mean for them, and what they need to do to avoid suffering the consequences of ignoring the call of reality.
In the mean time the MDR and IVDR implementation roll-out rolls on and the transition period is ticking away – 26 May 2020 for MDR and 26 May for 2022 for IVDR. Yet, I still encounter management reviews that say “there is this MDR /IVDR, we have this person that is thinking about it, a plan will result, and we have until 2024 anyway, we will start doing something when we’re finished with MDSAP / ISO 13485:2016 / whatever”.
Are those dates in 2020 (MDR) and 2022 (IVDR) firmly between the ears of your whole organisation – including your management? Because it’s only core business, and a situation of perish or not. Sounds exaggerated? Well, its only about your company’s core business for the whole EU and other CE mark dependent markets. How could that be significant, right? Maybe consider bringing in Batman to set your management straight. You can be the good cop and Batman will be, well, Batman.
Just as a recap of things I have said often on this blog already, here is a presentation I did yesterday for an audience at the Dutch Standardisation Institute (NEN):
Show this to you management and tell them they are not Chuck Norris – only Chuck Norris can be a kamikaze more than once.
First MDCG – stakeholders meeting
In the mean time we’ve had the first stakeholders meeting with the freshly set up MDCG and stakeholders on 5 March. This meeting gave us a lot of interesting information about where things are and where they are going.
How will the MDCG work?
The MDR and IVDR attributes a lot of competence to the MDCG, which is composed of the delegates of the member states presided by the Commission. The MDCG will sit on top of a structure of groups much like the current MDEGs, which are currently transferred to the new structure. The MDCG groups are currently going full pull in the roll-out of the CAMD roadmap. The CAMD has also produced helpful FAQs for the MDR and the IVDR in the mean time – I recommend you check these out as they do actually answer questions . Also, stay tuned because I understand that the FAQs will be updated soon.
The MDCG structure is in the process of being set up and the existing structure of MDEG groups is being transferred to the new structure. Stakeholders will be invited to join all groups except those for market surveillance and joint audits in spring this year. A new cluster on new technologies and borderline issues is being set up.
Implementation of MDR and IVDR
Here is a non-exhaustive run down of implementation actions currently in the works:
- The draft implementing act for reprocessing of single use devices will be made available for consultation soon. Highly relevant for any company that provides services of reprocessing;
- A new draft implementing act on Annex XVI products (‘non-medical devices’) will be developed in the next months as the member states could not agree on the previous version;
- Common Specifications for MDR will primarily be drafted by competent authorities (contrary to IVDs, where the stakeholders are in the lead);
- Common Specifications under the MDR and IVDR will take ISO and IEC standards into account but will not copy-paste them and will be made available for consultation at the end of the drafting process; and
- The CAMD implementation task force is working on more FAQs in addition to the first batch published earlier.The CAMD will set up a website where stakeholders can submit questions, which will be answered by a special inquiries group. This is expected to go live end of April.
Eudamed
Eudamed will be the database of databases for the EU medical devices and IVD system. The one database to rule and bind the other databases so to speak. You will not be able to place devices on the market in the EU without engaging with Eudamed, which is why this is an important project. The Eudamed roll-out is on schedule and looks to unfold along these lines:
- May 2018 – The first functional testing starts – actors module is first (because best defined in MDR), devices module next shortly after
- 25 May 2018 – MDCG accepts all functional specifications (as per article 34 (1) MDR)
- Autumn 2018 – Eudamed WG comes with detailed functional specs for Eudamed so companies can develop interfaces
- Q1 2019 – Further functional testing
- May 2019 – Public Eudamed site goes online
- September 2019 – Eudamed is ready for the formal audit (article 34 (2) MDR)
- March 2020 – Eudamed goes live and notice in OJ (article 34 (1) MDR)
One of the big dependencies for Eudamed is the national process for assigning Single Registration Numbers (SRNs) to economic operators by the member states. You need an SRN for Eudamed to be able to recognize you. Assignment of SRNs are national processes that different member states must set up independent of each other, so companies must check in which member state they must request their SRN for which capacity of economic operator (manufacturer, authorised representative, etc.). For example, the Netherlands is now working on deciding how to do this and whether the government entity that manages the current class I devices and IVDs notification database should do this or not.
Joint assessments of notified bodies
The Commission and competent authorities informed that they feel they have made sufficient resources available for a timely joint assessment of all the notified bodies that handed in a timely application. However, they cannot guarantee anything regarding the time it will take notified bodies to correct the non-conformities found in the joint audits. So far they have experienced bottleneck issues not on their side but rather on the side of notified bodies, e.g. with meetings being scheduled late because lack of capacity on the part of notified bodies.
Notified bodies also seem (this is my observation) difficulties to maintain the personnel necessary for the scope that they applied for. In some cases just one person quitting pending the audit process can mean that the notified body is unable to qualify for the full scope that it applied for. This something you should closely monitor your notified body for. Does your device need a scope that is out of the ordinary (animal tissue, combination product, etc)? Then make sure that you monitor over time that your notified body applied for the required scope and keeps the people required for that scope.
Notified bodies have been told that they cannot consult, which means also not on what they expect the certification requirements to be under the MDR and IVDR, so no offering of MDR and IVDR pre-certification programs. This makes it more than extraordinarily important for companies to rely on excellent internal staff (if you have them, treat them well because they are worth their weight in gold) or excellent external advice (if you can find them, because consultants that still have capacity because they are just starting work on MDR and IVDR things are probably not the best).
The Commission planning is that the first notified bodies will be notified half 2019. Notification will take place on a first finished first notified basis.
In the mean time I have found that notified bodies are in disarray about what they can say and what they cannot say about their accreditation process (which is actually more than they often think or are willing to admit for competitive reasons), and for that reason sometimes put on a show of ‘all will be well’ or just completely clam up (and everything along that spectrum), or mislead by stating ‘intentions’ about scope and time of application and then not follow through. So be vigilant and take no bullshit from your notified body – in the end notified bodies are companies and they will always put their own interests before yours.
UDI
As you know, even though the MDR and IVDR contain quite a lot of detail on UDI, most detail is still to follow and an implementing act on UDI issuing entities is under development. Guidance on UDI is to be published this month.
Harmonisation
The Commission expects about 230 standards to be harmonised under the MDR, for each of which a mandate is needed, most of which are existing harmonised standards under the MDD and AIMDD. These mandates for the MDR and IVDR have not been given so far.
The Commission still has a love-hate relationship with international standards, because harmonisation remains a matter of doing gap assessments agains the MDR and IVDR essential safety and performance requirements. All ZA, ZB an ZC annexes to harmonised standards will need to be amended to the MDR and IVDR as part of this effort. The Commission has indicated it will focus on processing the horizontal standards for the moment.
We will need to see how the Commission will act in common specifications (which under the MDR and IVDR can fulfill the role of standards) given the slow harmonisation process.
Corrigendum to MDR and IVDR
I understand that the European Commission is working on a corrigendum to the official texts that will be published in this year – there are little inconsistencies and small points here and there. This text will replace the current official texts that were published in the Official Journal on 5 May 2017. Expect no re-opening of the text – that would require another loop through the legislative process.
Work on vigilance
It was reported that the revision of the vigilance MEDDEV (2/12/1 rev 8) is ongoing. Templates for vigilance reporting into Eudamed will be finalized by October 2018 and then implemented into Eudamed. The Manufacturer Incident Report (MIR) form will be published in May 2018.
Clinical evaluation
The million Euro (or more even, depending on the product) is “when do you have sufficient clinical evidence?”. The good news is that the CAMD is are working on guidance on clinical evidence and equivalence, as announced in the Roadmap.
Interestingly, and contrary to rumors that I picked up before, MEDDEV 2.7/1 Rev 4 on clinical evaluation will not be amended anymore before the MDR date of application.
Breaking Brexit Batman!
 And then there is news on Brexit: a hard Brexit has been averted by the UK agreeing to a 21 month transitional period during which EU legislation will continue to apply in the UK with the UK having no influence over its creation (“leave Britain essentially a non-voting EU member until the end of 2020 to ease concerns for business“). In other words, the UK will be a kind of Switzerland from April 2019 to December 2020: bound by EU medical devices law with no formal influence over its creation. As a result, the dire situation painted by the Commission in its notice about the Brexit consequences in the context of CE marked goods is temporarily postponed. P-O-S-T-P-O-N-E-D, mind you, not averted. While the transitional period shows that the UK government has made remarkable steps in aligning with the realities of international trade policy, it is still not sure what the post-Brexit situation looks like. Actually, it’s not even certain at this moment what the situation will be regarding medical devices as there is no explicit confirmation at this point that the transitional period will mean business as usual for the medical devices system.
And then there is news on Brexit: a hard Brexit has been averted by the UK agreeing to a 21 month transitional period during which EU legislation will continue to apply in the UK with the UK having no influence over its creation (“leave Britain essentially a non-voting EU member until the end of 2020 to ease concerns for business“). In other words, the UK will be a kind of Switzerland from April 2019 to December 2020: bound by EU medical devices law with no formal influence over its creation. As a result, the dire situation painted by the Commission in its notice about the Brexit consequences in the context of CE marked goods is temporarily postponed. P-O-S-T-P-O-N-E-D, mind you, not averted. While the transitional period shows that the UK government has made remarkable steps in aligning with the realities of international trade policy, it is still not sure what the post-Brexit situation looks like. Actually, it’s not even certain at this moment what the situation will be regarding medical devices as there is no explicit confirmation at this point that the transitional period will mean business as usual for the medical devices system.
Also, member states governments are unable to positively confirm this at this moment, so this will need to be fleshed out more explicitly for the transition period. I’m saying this because the current draft Article 50 agreement (the agreement that is being negotiated for the UK’s withdrawal) is still firmly vague on whether the UK will have notified bodies that have any competence for the EU (like the Swiss currently have) – see article 42 of that draft agreement. Therefore, it is by no means a situation in which the devices world can now comfortably sit on its hands pending all of this.
More clarity to follow, hopefully. But don’t sell the fur before the bear has been shot, as we say in Holland, even after we exterminated our last local bear centuries ago.
National implementing legislation
Finally, companies should make sure that they are informed of the implementing legislation that the member states are working on currently. The MDR and IVDR leave lots of points for national discretion of implementation, and judging from for example the draft for the implementing act that the Netherlands is preparing I can say that member states like to go all out in their options because for them it’s a nice opportunity to recalibrate national policy.
An IVDR seminar specially for the IVD industry
I have noticed that there is a dearth of information for the IVD industry and that this may lead to many (especially smaller and non-EU IVD companies starting their implementation too late to be ready in time), so I decided to organise an IVDR specific event at my firm’s office in Amsterdam on 18 April, in the seminar format that you are used to from Axon Lawyers.
We will give specific attention to the CAMD Roadmap items for the IVDR:
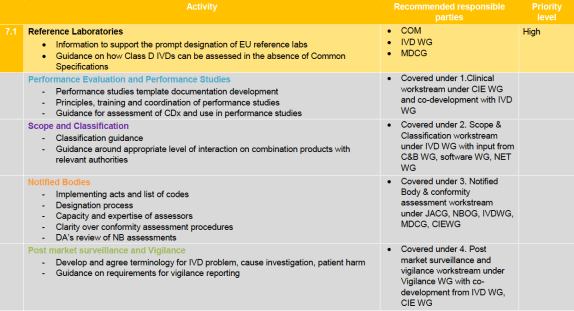
It looks like we may have an additional focus on the dangerous substances regulation for IVDs and medical devices in general, so if you’re interested in that, this is your chance.
We have secured the participation of Anja Wiersma of mi-CE (who knows EU IVD regulation like no-one else and is president of the RAPS Netherlands chapter) and a surprise industry speaker from the IVD industry.
So if you are in IVDs and have interests in the EU market you should attend – register here. It’s free and you can come with as many people as you like (just let us know how many). We’re not streaming or recording the seminar, because we believe that the best transmission of expertise takes place on a person to person basis. As usual we will seek permission of the other speakers to be able to publish all presentations on our website.




Hi Eric
Would you mind if we republish this on our site? Such useful insight!
Kathryn
Hi Kathryn, of course – you are most welcome. Please attribute source and link to my blog. Best regards, Erik