 Everybody has been in enormous suspense about how the second corrigendum to the MDR and IVDR would turn out. The version that came out of the European Parliament’s ENVI committee vote contains a number of very technical points that I will not elaborate on. But it also contains the big break for certain class I devices, the ones that are either up-classified under the MDR (and need a CE certificate under the MDR as a result) and the re-usable surgical instruments (which need a CE certificate for the reusability aspects under the MDR).
Everybody has been in enormous suspense about how the second corrigendum to the MDR and IVDR would turn out. The version that came out of the European Parliament’s ENVI committee vote contains a number of very technical points that I will not elaborate on. But it also contains the big break for certain class I devices, the ones that are either up-classified under the MDR (and need a CE certificate under the MDR as a result) and the re-usable surgical instruments (which need a CE certificate for the reusability aspects under the MDR).
At this moment, the corrigendum has not been finally approved. This is supposed to happen somewhere before Christmas, so the class I devices industry may get a nice Christmas present under the tree – or not. If the corrigendum is still shot down on the finish line, this will be too bad as most of the class I devices that are unclassified under the MDR will likely not be able to find a notified body in time and will need to temporarily or permanently leave the market.
Class I MDR only, not IVDR
It concerns MDR devices only? Yes, this corrigendum does not contain any provisions with regard to up-classified IVDs, which comprises basically 85% of all IVDs currently on the market. This is why IVD manufacturers should still go full speed ahead with their IVDR implementation. I think it is unlikley that there will be a change in transitional regime for this large a group under the IVDR, because that would essentially change the nature of the transitional regime. But what do I know? It might still happen if the bottleneck for the IVDR is as severe as I predicted.
In the below presentation I have set out the consequences for class I devices of the corrigendum as I see them currently. I explain them in person in a lot more detail on the Medical Device Made Easy podcast about this exact subject (this is the first part of two parts about class I devices and the MDR, next one to follow later). I recommend that you follow this podcast for everything MDR and IVDr related, because like this blog that series of podcasts is starting to form a very nice body of knowledge and training material on EU devices regulation under the MDR and IVDR and provides a lot of expert knowledge on it. And it is very very practical.
The corrigendum creates two classes of class I devices:
- the ones subject to the corrigendum and thus included in the article 120 (3) and (4) MDR regime; and
- the ones for which it is business as usual and that must meet the full MDR requirements by the date of application (26 May 2020).
Subject to corrigendum devices (“upclassifieds”)

The class I devices subject to the corrigendum are all devices that are up-classified under the MDR under Annex VIII, notably software (rule 11), devices with nanomaterial (rule 19), inhalers (rule 20) and substance based devices (rule 21) and the re-usable surgical instruments. These devices are subject to the transitional regimes in article 120 (3) and (4) MDR provided that they have a valid declaration of conformity before 26 May 2020. They can then
- Be placed on the market under that declaration of conformity until 26 May 2024 – provided that there is no signficant change to intended purpose or design (more about significant changes below); and
- Be sold to end users until 27 May 2025 (so the devices placed on the market before 26 may 2024 have another year to make their way to the end user).
And they must be covered by an MDR CE certificate that a notified body must issue by 26 May 2024, in order to continue to be placed on the market after that date.
See in the embedded presentation above what these devices must do under article 120 (3) MDR, and all of that is minus Eudamed for the moment.
Word of advice: some clients of mine in this boat are already saying “Yay! We can postpone our CE certification for the MDR until 2024!” and I am telling them “No, unless you change the name of your MDR project into Project Kamikaze and put a note in your calendar for 1 April 2024 saying ‘Erik told us in 2019 that this would be a bad idea and now we owe him a bottle of good champagne.’.”.
Why is it a bad idea? By the end of the article 120 (3) MDR soft transition period everybody and their mother, the extended family and all cousins too need a CE certificate as well. Not only will there be the normal MDR certificates demand at the level of normal market conditions, but also all AIDD and MDD certified devices that need MDR certificates by then. And the difference is: these manufacturers will already be existing customers of the notified bodies, so they will go first and even for them it is not sure that there will be enough notified body capacity by then. So if you wait until the end of the transition period you will be lucky if a notified body will even pick up the phone by then to tell you that they are not accepting new customers. Better plan your inevitable transition to an MDR certificate way before 2024, preferably 2021 or 2022 as this will likely be a less busy period. Planning it at the last moment possible is a recipe for trouble – you’ll have no one to blame except yourself when you wait until the last moment to find out that all the notified body capacity is spoken for. I will quote you Kant, one of my favorite philosophers: “Sapere Aude” (‘dare to be wise’).
Signifcant change
The significant change under article 120 (3) MDR is kind of a landmine of a concept that can really mess things up for you as a class I manufacturer. That’s why it is really important to know what a significant change is. Fortunately there is guidance on the way! Oh, wait…:
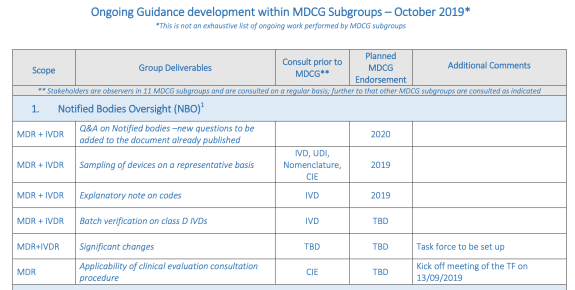
Yes, you are seeing this right. In the last version of the ongoing guidance development document the task force for this (the concept has somewhat of a sense of urgency to it) still had to be set up and planned release is TBD. How is that for a sense of urgency?
The consequence of a significant change is that your declaration of conformity will not be valid for the article 120 (3) regime anymore, which means your CE mark is instantly invalid, causing non compliance for every every next device that you place on the market.
But things can be even worse if the significant change affects the installed base of devices already at customers. A totally non-hypothetical scenario that I am sure will happen is that of a software update to a manufacturer’s software installed in hospitals turns out to be a significant change, e.g. because it is a line extension of the installed software, adding addition convenient functionality or is a fundamental redesign for the software to be able to run on 64 bit operating systems too. Especially in the last case the IT department may roll-out the update without thinking because nothing changes. In both cases releasing the update will constitute a significant change. In order to help you decide if a change to software is a significant change LNE/G-MED’s guidance on significant change has a helpful flowchart (no 5) in it for software. The guidance has other helpful flowcharts too. But, this is what LNE/G-MED thinks, and as class I device manufacturer you are dealing with competent authorities and not with notified bodies.
Full MDR by 26 May 2020 devices

The devices that are not subject to the corrigendum are going to have to be fully compliant by 26 May 2020. These device are subject to the full MDR (minus Eudamed for the moment).
This is a big step up for class I device manufacturers that have been operating under an MDD Annex VII QMS, because they are going to an ISO 13485:2016 plus QMS. Also, these manufacturers will run into the alternative meanings of CE and MDR: Clinical Evidence and More Data Really. You will not only need to redo all of your clinical evaluations, but you will need more data as input (which you will likely not have if you have not been doing active post market surveillance as many class I manufacturers have not been doing).
The above presentation provides an overview of what you can expect as a class I device manufacturer under the MDR. If you start to figure all of this out only now, you will have a very steep learning curve and a lot to do in very little time.
Suspense suspense
We’ll have to see if the corrigendum 2 is a done deal or not. If it is, it temporarily solves the predicament of the class I devices that are unclassified or need a reusability certificate under the MDR. But, it’s just kicking the can down the road – don’t forget that these devices need an MDR certificate by 26 May 2024, so better start with that sooner rather than later. And all the other class I devices: be prepared – you’re on for 26 May 2020.



Erik,
The corrigendum states that “…a device which is a class I device pursuant to Directive 93/42/EEC, for which the declaration of conformity was drawn up prior to 26 May 2020 and for which the conformity assessment procedure pursuant to this Regulation requires the involvement of a notified body…”
Does this mean that only class I devices which have an EC cert from an NB receive an extension, but not class I self-certified devices which are upclassified to Class IIa.
For example, a stand-alone software currently Class I would not get an extension, because it does not have an EC cert.
Hi Mike – no, it does not mean that. The CAMD MDR Q&A from 2017 already said that class I devices with certificate (Class I m and s) could use article 120 (3). The corrigendum 2 now means that all currently class I devices that would be up-classified under the MDR can continue under their pre -26 May 2020 declaration of conformity without needing a CE certificate, unless a significant change is implemented (which in the case of software has a low trigger, see the LNE/GMED guidance I’ve referenced on the blog).
OK this is a lengthy comment – I’m looking for confirmation of my understanding:
Any Declarations of Conformity (new or revised) issued after 26 May 2020 are subject to the EU MDR.
• Any Class 2a devices already on an MDD Declaration of Conformity can continue to be sold under that DOC
• Any new or significantly modified devices that would require the manufacturer to issue a new DOC is/are subject to the EU MDR (not the MDD)
• Class 1 products that are “self-declared” can be issued an EU MDR Declaration of Conformity, providing that we have the documentation to support the EU MDR requirements.
The same caveats apply
• May be placed on the market under the MDD Declaration of Conformity until 26 May 2024 – provided that there is no significant change to intended purpose or device and
• Be sold to end users until 27 May 2025 (so the devices placed on the market before 26 May 2024 have another year to make their way to the end user).
Is it correct to say that companies that have not been able to schedule/pass an EU MDR audit cannot launch new products in the EU after 26 May 2020 (except if we are compliant to the EU MDR for Class 1 products)?
Hi Mara, your understanding is not correct on some points, and we’ve had the corrigendum 2 adopted in the mean time that changes things for devices that are class I now and higher under the MDR. If you’d like my formal opinion, feel free to drop me an email at my work address erik.vollebregt@axonlawyers.com and we can discuss this further.
Dear Erik,
Risk Class I devices legally placed on the market with the importer before May 26 2020 fully compliant with the MDD can continue to be put into service in the EU until when?
That would be possible until 27 May 2025.