 Welcome to 2020 – happy new year everyone. I don’t care if your conventions do not allow me to wish you best wishes after a certain date anymore. I wish you a happy 2020 because I genuinely hope you will have it. If you are a reader of this blog and you have a happy 2020, it will mean that a lot more went right in the MDR roll-out than I would expect based on the outlook at this moment.
Welcome to 2020 – happy new year everyone. I don’t care if your conventions do not allow me to wish you best wishes after a certain date anymore. I wish you a happy 2020 because I genuinely hope you will have it. If you are a reader of this blog and you have a happy 2020, it will mean that a lot more went right in the MDR roll-out than I would expect based on the outlook at this moment.
Over the Christmas holidays I wrote my contribution to the Dutch Health Law Association’s 2020 periodical book discussing developments in healthcare regulation (in case you read Dutch: Preadvies van de Vereniging Gezondheidsrecht), which is about medical devices law this year. It will be published spring 2020 and my conclusion is not different from my not so happy Christmas carol post just during the Christmas holidays 2019 (which I wrote as I was writing the contribution to the book).
The reality of things
 The reality of things is that things are heading to an urgent critical mass (or mess, pun intended), because there is less and less time to make a meaningful difference.
The reality of things is that things are heading to an urgent critical mass (or mess, pun intended), because there is less and less time to make a meaningful difference.
We have about five months to go to the date of application of the MDR. Five is not much. It’s the amount of fingers on your hand – I’ll borrow you mine to make the point. See: only five fingers. Not that many. A fast typical conformity assessment under normal circumstances takes at least six months. Six is more than five. This means that even theoretically no additional notified body added in Q1 2020 will make a a difference before May 2020.
When I wrote the Not so happy Christmas carol post last December some people thought I was being overly negative and I even started believing that myself at some point.
But then I resumed work after the holidays and the reality of the devices world washed over me again with clients asking my advice in an avalanche of problems and with signals that I see in the market:
- clients facing massive delays in any interaction with notified bodies;
- clients approaching notified bodies for the first time are not able to find any notified body that will onboard them;
- clients facing notified bodies ‘offering solutions’ that will costs them double or more just for the notified body to renew a certificate in time for it not to expire;
- notified bodies ‘notifiedbodysplaining’ how their capacity is tied up to the extent that they are unable to make any predictions about certification decisions, not for (AI)MDD recertification and not at all about MDR certification;
- health institutions being mostly oblivious about what the MDR and the IVDR will mean for them (or in vicious state of denial about the in-house production regime);
- class I manufacturers mostly having absolutely no idea what the MDR will mean for them and not understand at all that there is a large gap to cross;
- independent distributors far and wide not wanting to touch the MDR with a 10 meter pole (yes, that’s more than 10 feet as the expression is originally phrased) and distributors calling themselves ‘wholesalers’ flat out denying that they are in scope of the MDR and IVDR, even though they evidently are;
And the list goes on. My pessimism is back.
The system is not antifragile and has an agency problem
 We ended 2019 with the new Commissioner for Health jedi mindwaving the member states about the status of Eudamed and notified bodies and what that means for availability of medical devices on the EU market. Nobody seemed willing to admit publicly that the EU regulatory system for medical devices has not been designed to deal with the enormous spike in notified body and authority capacity needed for the transition to the MDR and to the IVDR. The system can just about operate at nominal capacity in its fragile equilibrium of structural under-resourcing, but the MDR and IVDR did nothing to make the system antifragile (i.e. capable of becoming sufficiently robust under stress) enough to deal with the efforts required for the enormous bulge in the pipeline resulting from the fact that all notified bodies needed a new accreditation and all certificates need to be re-issued under new criteria. In essence the MDR and IVDR have been set up as a massive agency problem waiting to happen, and that’s exactly how it unfolded, or in the words of Nassim Nicholas Taleb describing the agency problem:
We ended 2019 with the new Commissioner for Health jedi mindwaving the member states about the status of Eudamed and notified bodies and what that means for availability of medical devices on the EU market. Nobody seemed willing to admit publicly that the EU regulatory system for medical devices has not been designed to deal with the enormous spike in notified body and authority capacity needed for the transition to the MDR and to the IVDR. The system can just about operate at nominal capacity in its fragile equilibrium of structural under-resourcing, but the MDR and IVDR did nothing to make the system antifragile (i.e. capable of becoming sufficiently robust under stress) enough to deal with the efforts required for the enormous bulge in the pipeline resulting from the fact that all notified bodies needed a new accreditation and all certificates need to be re-issued under new criteria. In essence the MDR and IVDR have been set up as a massive agency problem waiting to happen, and that’s exactly how it unfolded, or in the words of Nassim Nicholas Taleb describing the agency problem:
“Situation in which the manager of a business is not the true owner, so he follows a strategy that cosmetically seems to be sound, but in a hidden way benefits him and makes him antifragile at the expense (fragility) of the true owners or society. When he is right, he collects large benefits; when he is wrong, others pay the price. Typically this problem leads to fragility, as it is easy to hide risks. It also affects politicians and academics. A major source of fragility.”
It also affects politicians – and indeed it does for the MDR and IVDR.
The lack of capacity is being felt keenly an acutely by stakeholders, and is mostly managed badly for a lack of options and resources. I have come up with a thought experiment that I would encourage everybody (and especially authorities) to apply to any situation MDR or IVDR related to expose the agency problem. Ask yourself in every situation: “Would we be OK with medicines agencies operating this way?”. Think about it. In many situations ‘the world would be too small’ (paraphrased Dutch expression) because it would be inconceivable in relation to medicines that the system or the government agency responsible operates this way. Notified bodies, however, are almost never government agencies, but they do exercise government authority. Some examples I have come across, for your consideration:
- After accepting your application the registration authority says it will double registration fees and otherwise not finish your application process in time for your existing authorization to expire.
- The registration authority says it is not going to finish your application in the time frame promised, because it can actually not promise anything because it’s so busy and literally tells you that if you don’t like it, you are welcome to go to another agency.
Would we like the EMA or the national medicines agencies to operate this way? Hands up if we do, and please send in a comment if you think this is a good idea so we can propose amendments to basic principles of good administrative practice.
The bottlenecks summed up (again)
 The reality of things is that even if the Commission is emphasizing the small successes (yay, another notified body accredited or look here, another guidance document published) and the Member States are doing their absolute utmost to trust the Commission when it is totally Jedi mind waving them, they’re not fooling anyone else.
The reality of things is that even if the Commission is emphasizing the small successes (yay, another notified body accredited or look here, another guidance document published) and the Member States are doing their absolute utmost to trust the Commission when it is totally Jedi mind waving them, they’re not fooling anyone else.
MedTech Europe published a very clear and accurate paper that sums up all the bottlenecks, and shows that each of the possible ways to have devices on the market post May 2020 (an MDR certificate, a renewed (AI)MDD certificate or a national/Union-wide exemption) is affected one way or the other by the critical lack of capacity. They also point to potential solutions for each bottleneck, which I am not repeating here – please read the paper, it’s important. Spoiler alert: most of the solutions are to deliver as soon as possible on what should have been delivered years ago. This is also not new, but more urgent than ever.
I will summarise the bottlenecks (again) and invite you to read the MedTech Europe paper for the solutions.
MDR certificates
There are insufficient MDR notified bodies accredited to make the difference, essential guidance is lacking and the system is not ready in many respects and for certain devices. It takes notified bodies considerable time to get up to full accreditation speed after they have been accredited and even under the best of circumstances it takes at least half a year to process a conformity assessment application. The fact that a notified body has been accredited does not mean that it will be able to make the difference before 26 May 2020. The notified bodies accredited in Q1 2020 (including the remaining eight promised for 2019) will likely not issue a single certificate before the date of application of the MDR.
Renewed (AI)MDD certificates
There is not even enough capacity to renew all the existing certificates for the 2020-2024 grace period, and there is complete unclarity about what the duties of notified bodies overseeing these certificates past May 2020 are. As things stand, they can give any manufacturer three months notice, leaving the manufacturer in a situation where it will never ever be able to have an MDR certificate at another notified body (too busy, and takes (much) more than three months) and it is uncertain whether the orphaning procedure in article 46 MDR applies only to MDR certificates or also to (AI)MDD certificates valid past May 2020 based on article 120 (3) MDR. And there is still the uncertainty around the interpretation of the concept of significant change, which can invalidate certificates just like that. The fact that the guidance promised by the CAMD in December 2017 is still not here is quite frankly astounding.
Also, there is (still) no solution for notified bodies not renewing a certificate in time, or refuse to accept the renewal application.
National and EU exemptions
As under the old directives the primate of exemptions to CE marking remains with the Member States under the MDR. There is a possibility for extending a national exemption to the whole Union territory pursuant to article 59 (3) MDR – which means that there must first be a national exemption that applies to one Member State. Each member state may use their own criteria for that, which often involve that the exemption is the exception, i.e. not intended as a general alternative market access mechanism for everyone that could benefit from it. You will also often need to show that you are on board with a notified body already, which is kind of problematic because most are not accepting new customers at the moment.
Subsequent extension of the exemption to the whole Union (so the manufacturer does not have to make an application in each Member State) based on article 59 MDR is limited to ‘exceptional cases relating to public health or patient safety or health’, so not intended as a general exemption for everyone in trouble it would seem. And each exemption needs its own implementing act under the MDR to be adopted under the examination procedure, which looks like this:
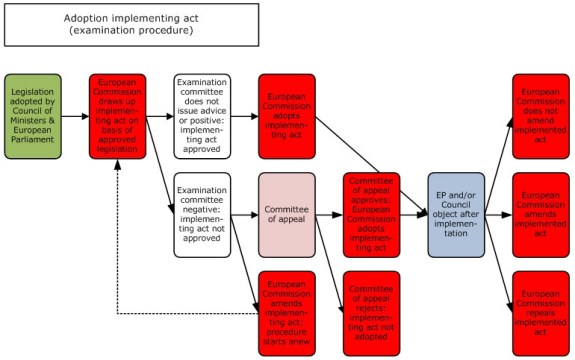
As you can see, not complicated and slow at all – not. Quickly change the MDR for a more streamlined and quicker process perhaps? Not enough time to do this before May 2020.
Furthermore, it is not clear whether it is allowed for devices exempted this way to affix (or keep affixed) CE marking. If this is not allowed, this will severely impact exports to third countries that allow devices on their markets based on CE marking.
You finally really seem to be doing it
 One of the most powerful images that I can come up to underline the sense of urgency of this all is the final scene of the first Planet of the Apes movie.
One of the most powerful images that I can come up to underline the sense of urgency of this all is the final scene of the first Planet of the Apes movie.
Charles Heston’s character realizes at the end of the movie that the barren and primitive place in which he found himself the whole movie was in fact the post-apocalyptic earth. Would we like a health system just as barren as that of the planet of the apes? Maybe watch the movie for yourself to get an impression but (spoiler alert): the planet of the apes is missing a lot of nice to have and innovative medical technology that used to be available because it is kind of primitive. Of course this is a dramatic example, but I am running out of ways to express the urgency of the problem.
MedTech Europe politely captions its paper with “A Call to Action” because they have to be polite. I have less constraints and will be less polity because I am a (potential) patient myself, I have family members that depend on medical technology for their life, and others for their quality of life. And I am truly upset and disappointed about what we have to show for the MDR implementation at this moment (and don’t even get me started about the IVDR). I would like the best regulatory system so everyone can have access to the best and most innovative medical technology, not to the bare necessities only because we could not be bothered to make the regulatory system work.
Of course in the end the politicians will say it is nobody’s fault (agency problem anyone?), except that this would be wrong. The writing of things coming to this critical mass has been on the wall for a very long time, just read back this blog three years.
Let’s realize that we are all (potential) patients and want the best for our loved ones, ourselves and why not society at large, and make this thing work by adding clearly needed antifragility now that there still is some time. All the solutions are there, we know them, MedTech Europe helpfully repeated them. And the rest is politics.



Excellent summary of the current situation by Erik !! It is sometimes ambarrising to see how politicians move around and try to implement the regulation in Europe.
I am happy that many Industry partners are preparing despite the many uncertanties and hurdles to take because their prime concern is still the safety and quality of life of many patients around the globe including the EU.
I’m afraid Erik Vollebregt is right in his dystopic view of the current situation for the MDR. I’d like to expand a bit on the problem with the “not fully functioning” Eudamed. Eudamed is, in my opinion, the backbone of the entire MDR construction. Nothing much is going to work as planned without this envisioned database.
On top of making the fundamentals of Eudamed work, article 33 in the MDR says:
“When designing Eudamed the Commission shall give due consideration to compatibility with national databases and national web-interfaces to allow for import and export of data”.
Considering the different, and continuously changing, systems used by Competent Authorities in our member states I suggest the above article 33 promise must give the poor code-writers in the Commission the worst possible nightmares.
Let’s hope that the Eudamed does not turn out to be just (another) a pie in the sky.
Lennart Philipson, Sweden
Former regulator, now a consultant
Dear Erik, thanks much for this so well formulated summary of the situation. This is to the point and we can confirm exactly what you describe for each of the points.
Best
Michael
And where are the harmonized standards for the MDR? The current harmonized standards lists in the Official Journal for the directives have not been updated in years. These stale lists no longer reflect the “current state of the art” required by the MDR, as several key standards have been updated since the last OJ update. We are spending a lot of time doing gap analyses on revised standards to justify their use.
And the only status from MDCG on Common Specifications development is “TBD”.
Standards are important compilations of lessons learned, sometimes learned at the expense of patient lives. Not having up-to-date harmonized standards is a safety problem that needs to be addressed STAT.
Erik, may I ask where you got this statistic: “A fast typical conformity assessment under normal circumstances takes at least six months”?
The only MDR timeframes to which I am privy are those associated with the first MDR certificates issued by BSI UK and TUV SUD:
BSI UK issued its first certificate a little over 200 days after MDR designation. I’m inclined to think this process may have been slowed a bit by its being the first (things to sort out that had not yet been) and with the distraction of Brexit.
TUV SUD issued its first certification less than 100 days after MDR designation, for a Class III device.
Of course, these were not “normal” circumstances, but I don’t think enough NBs have been certified long enough for anyone to know what “normal” circumstances are going to be. If this refers to “normal” MDD circumstances, then I’m not sure how generalizable that experience is to the current, as-yet-to-be-seen “normal.”
Hi Julie, that statistic comes from the MedTech Europe report referenced in that blog. The first certificates issued are of course no measure for normal procedure duration as these are proofs of concept.
Indeed, and to elaborate a bit further: Our members describe the ‘typical’ certification timeframe (under the MDD, and before today’s capacity constraints became so bad) as “3-9 months,” depending on the device’s level of risk and novelty.
That’s the figure we tended to use last year, e.g., in open letters to European Commissioners.
For simplicity’s sake, our latest Call to Action, which Erik references, simply refers to “6 months” as the middle-point in this 3-9 month range.
Erik, thanks. They are proofs of something, for sure. 🙂
Oliver, thanks for the clarification. What do your members use as the starting point for the 3-9 months?
If I understand this correctly now, I think this data source isn’t consistent with “typical fast conformity assessment”? It is instead the average conformity assessment? (Mean, median, mode, won’t even go there…) If not caveated with “average,” then it takes at least 3 months, not 6 months?
Unless there is a process called a “fast conformity assessment, which all of your members were referring to, and therefore there is another, “not-fast assessment procedure” that is not included in the 3-9 months and presumably would take longer?
(I realize Erik is a lawyer trying to make a point, not a data analyst, but I’m a data geek who is fascinated by this whole saga and want to be sure I’m understanding these things correctly.)