 As we are just past the halfway point of the MDR transitional period for the MDR and are well into the one for the IVDR there is one thing that has become very very clear: the transitional period in the MDR and likely the one in the IVDR is not actually a transitional period for industry and certainly not the one you are looking for. It is more like an extended implementation period for the authorities to complete the regulations that came out of the legislative procedure as finished regulations but as a half-baked regulatory system that required a lot of additional implementation for it to be functional.
As we are just past the halfway point of the MDR transitional period for the MDR and are well into the one for the IVDR there is one thing that has become very very clear: the transitional period in the MDR and likely the one in the IVDR is not actually a transitional period for industry and certainly not the one you are looking for. It is more like an extended implementation period for the authorities to complete the regulations that came out of the legislative procedure as finished regulations but as a half-baked regulatory system that required a lot of additional implementation for it to be functional.
As reported in previous blogs, the implementation machine at EU level (which is mostly a concerted effort by national authorities that have a difficult time agreeing on the details of implementation of the CAMD Roadmap). We were initially happy with the prioritization of items in the roadmap, until they unfortunately turned out to be aspirational rather than committal.
Rolling plan
The European Commission has now released a working plan for the remainder of the transitional period, the “rolling plan” with expected timelines and next steps for implementation of items from the MDR – unfortunately also rather non committal.
As you can see in the rolling plan document, most of the things will happen Q4 2019 / Q1 2020, in other words: critical implementation mass at the end of the transitional period. This is an issue for manufacturers seeking to implement the MDR and IVDR, because this means that although the framework of the MDR and IVDR was ready by May 2017, with most of the sometimes very critical details to prepare for to come much later, leaving essentially no meaningful implementation period for industry.
The implementation period is effectively used by the authorities for their own roll-out of the system, rather than to provide industry with a meaningful period to transition each and every device on the EU market to the new system, which is already a Herculian effort with a full three years available.
Eudamed and Single Registration Numbers
A poignant example are the Eudamed Single Registration Number (SRN) process and interfacing specifications (not even starting about Eudamed’s ready for use date as such). SRNs are the only thing that the EU does not provide – you have to get them on a national level and the member states have to set up mechanisms for issuing them after verification of the identity of the manufacturer, authorised representative or importer.
If you are a company with many products, you need to be able to talk to Eudamed on a machine to machine basis as soon as Eudamed goes online. Without an SRN however it seems you cannot interact with the Eudamed database, which will be necessary when Eudamed becomes applicable.
Have you checked with the competent authority in the member state where you are as manufacturer or where your authorized representative is when they will have an SRN for you and what you need to do for that?
CS for non-devices
Another one is the common specificiations for non-medical devices: arriving November 2019 while you should be compliant by 26 May 2020. That’s a cool six months to re-invent a product that is not a medical device as a medical device and really not a meaningful transitional period. Good luck with that!
Harmonised standards
And how about the harmonised standards for the MDR/IVDR? Anyone? The regulatory system under the directives was built on the assumption of conformity with the directives if you conformed to a harmonized standard. We are carrying over this system into the MDR and IVDR so these harmonised standards are kind of important if you want to declare or assess conformity with the general safety and performance requirements under the MDR and IVDR and rely on the presumption of conformity that meeting a harmonised standard affords as COCIR points out quite rightly.
As the rolling plan says: consultation of the member states on the scope of the first mandate is ongoing; expect a decision by Q1 2019. Wait what – first mandate? So there will be more? Yes, apparently we have quite a number of harmonised standards under the current directives, so it looks like the EU will harmonise standards under the MDR in tranches. When what why how? Your guess is as good as mine. I’ve heard that the plan is to start with the most important horizontal standards first. Which are? Surprise! Your guess is as good as mine.
Where is the European Parliament?
Yet, the Commission is playing ‘nice weather’/’these are not the droids you are looking for’ also in European Parliament by saying that everything is on track and that there is nothing to get all excited about. Well, I suppose you could say that even a burning runaway train that is going to arrive late and blow up the station is ‘on track’ in a sense of the literal meaning of the words.
Because where is our fiery European Parliament in all of this? It was so totally prepared during the legislative procedure to call industry by the worst of names (“single use labelling is like printing your own money“, remember?). There will be at least some notified bodies in time – sure, but will they have even close to the capacity requirements to deal with the workload coming their way (see below for more about that)? These are the important questions. An incomplete and under-resourced system is not going to deliver and a failed system is not a successful system. Our guardian of the citizens and its ENVI committee with an opinion about everything devices related seems to be comfortably sitting on its hands watching the European implementation machine fail to deliver a complete regulatory system timely.
The European Parliament will probably blame someone else because they had to make so many compromises to have the rules adopted in the first place. This is seems to be the approach that Ms Roth-Behrend (the wings are on fire again) seems to be saying the media push started by the ICIJ’s Implant Files: blame everybody but us. How convenient. But how about maybe taking some responsibility now?
Joint assessment process of notified bodies
The Commission also published an update of the joint assessment process of notified bodies. This nothing to become enthusiastic about, even though the Commission tried to put a positive spin on this in European Parliament by saying that more than half of the currently notified notified bodies have applied. Hurray! Does that mean that the capacity the market requires for transition all CE certificates on the market to the new system will be online timely?
Probably not: so far only 6 notified bodies have handed in CAPA plans (which likely means that all of the other applications have not made it to that level because as far as I know no notified body sailed through the joint assessments without CAPAs), only one final JAT opinion was issued for a lucky notified body that might be notified in Q1 2019 (provided that the final designating authority report is also issued) and no final designating authorities reports (which marks the ‘internal’ end of the designation process) have been issued. This means that in the most optimistic scenario notifications will start to trickle in one by one from somewhere in Q1 2019. It’s like I tell my kids over and over again: your homework is not finished until it’s done. And this stuff is so way not done yet.
Let’s take a realistic view at capacity coming online and capacity needed as Medtech Europe has been doing. It is becoming very clear now that several factors are brewing to create a perfect storm of undercapacity of the system as explained in the below MedTech Europe infographic:
- more and more notified bodies are closing down or not applying for MDR/IVDR notification;
- the Brexit is coming up with ~ 40% of the EU notified body capacity in the UK at risk (although this problem is temporarily delayed now – see below);
- fewer and fewer notified bodies do more of the work.
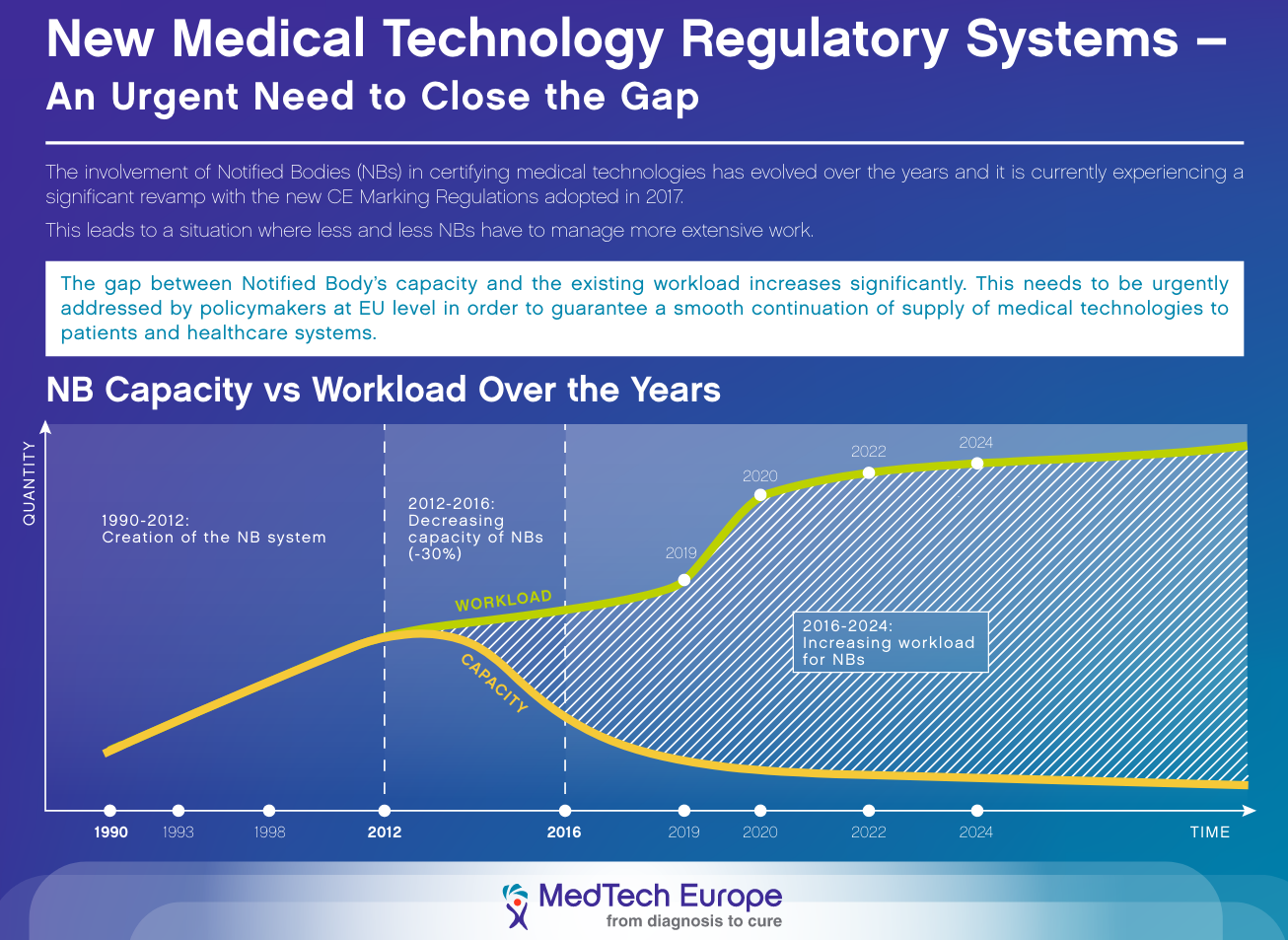
It’s not an exaggeration, this notified body capacity issue. In addition, notified bodies are increasing getting in worse shape, likely resulting from the pressure on their organization to get more work done than they can manage in parallel to the time consuming MDR and/or IVDR accreditation application.
Every week almost I am being contacted by desperate companies that run into catastrophic issues with their notified bodies (mainly the smaller ones), such as:
- certificate expiry because the notified body is unable to schedule and/or complete a timely recertification audit and often does not stick to agreed dates or promised actions, combined on occasion with the ‘solution’ that the deadline may be met if the client pays a considerably higher fee (I’ve seen twice normal rate);
- notified bodies flat out denying having received documentation as an excuse for missing auditing slots so they can skip the audit;
- notified bodies letting a certificate expire in a certificate in one of the above scenario and rather than taking remedial action charging the company for a full renewed entrance audit for the device and taking a very long time for that;
- suspending a certificate for all devices of the company while the intention was to suspend it only partially for one and then take weeks to reinstate the certificate;
- etc.
I think authorities can be a lot more sensitive to these kinds of things, regardless of whether they are caused by bad faith or negligence, or just by under-capacity, because manufacturers just have no meaningful legal recourse against a misbehaving or blundering notified body. Requesting specific performance under the certification agreement in court almost never works in practice, and internal dispute resolution mechanisms of the notified body will usually find that the notified body is right and take a long time.
So far I’ve found only one competent authority interested to hear about these things and willing to do something. Others are very standoffish or see it as commercial problem. That, I think, is way too easy if you’re finally responsible for the notified body’s good administration practices as notifying member state. Let’s not forget that the notified bodies are exercising delegated state authority. It would be very nice if the member states responsible would act like they are supervising their own institutions.
These notified body issues will only get worse as we get closer to the date of application of the MDR more notified bodies will keel over or start to exhibit this kind of behavior. Once we’re past the date of application this will really become a problem because then manufacturers cannot change notified body anymore. As long as they do not yet have their first CE certificate, it is unsure whether the MDR regime for delisted notified bodies in article 46 MDR applies and you can imagine how this will impact manufacturers that go for soft transition and then lose their notified body (and, as a consequence, their certificate).
Brexit
It seems that we have white smoke on the Brexit, but don’t get your hopes up yet. Whatever has been agreed (we still don’t know exactly) has to be ratified by a bunch of parliaments. Especially the Brexiteers in the UK parliament could seriously rain on this party.
But suppose the Brexit gets ratified. And then we have a transitional regime that lasts until 31 December 2020 – right in the soft transition period. And no certainty what comes next because that still needs to be agreed during the transitional period.
Implant Files
Then there are the Implant Files, a pretty large investigative journalism project aimed to hammer home the message that we have been hearing for years: CE marking sucks, notified bodies are evil corporations with a built in conflict of interest and government and industry are evil too, covering up piles of unnecessary cases of death and harm caused by faulty medical technology. Sounds dramatic, right? Because that’s news – no drama, no news that people are interested in.
But in my view this is no news at all – in fact this is all very much after the fact. We knew that the EU system could be improved, and that’s what’s happening with the new regulations. We also knew that vigilance reporting could be more transparent, which is why Eudamed is set up and will be much more transparent.
So here is my first reaction – since I’ve been quoted and have spoken to several journalists involved in multiple unsuccessful attempts to explain EU medical devices regulation to them, I feel I am entitled to an opinion in this matter. After all, journalists call me the manufacturers’ darling so I may have something to contribute to the discussion. For full disclosure: keep in mind that this is exactly what I am – manufacturers are my clients and I never make a secret of this so people know where I come from. I do not work for patients. So, believe what you will about facts but I do know the rules.
I think this initiative does underline that it is important what the changes in EU law with the new MDR and IVDR are going to do already: make more information about devices available to the public, such as by means of the Eudamed database that will contain a lot of extra information about devices. This is a good thing and yes, it could have been done earlier, but now it is at least happening.
However, I think the conspiracy theory assumption underlying many of the publications that this information is deliberately withheld is not productive, because this conspiracy does not exist in my experience. But these days you need a good conspiracy theory to have a good story so how do you create a conspiracy theory? The reporters tried to obtain massive amounts of vigilance reports relating to implants by use of freedom of information requests, because there is no EU law and (that I’m aware of) national law in the EU that allows for access to vigilance data held by authorities. However, vigilance reports are full of confidential technical information of companies about how a device works and sensitive personal data of patients and doctors that you cannot just make public like that. Freedom of information laws have protections built in to protect these interests. It would be kind of strange that anyone (anyone) can just petition authorities for documents containing technical information provided in confidence or personal data concerning health of patients, right? That’s however what they did, and then they were very angry that they did not get all the information they wanted immediately and without redactions in accordance with the law. Only one conclusion possible: it must be a conspiracy! I think that is somewhat disingenuous and frankly quite naive. But it’s good drama, so good news to sell.
I have read most of the German, Dutch, UK, French and Belgian publications that have come out so far and they paint a picture of evil government and evil industry withholding information and making bad devices that fail more often than they should. Nothing new so far – haters are going to hate no matter what and news without drama will not sell. I think that in reality the picture is far more nuanced than presented that but that would not be news of course. CE marking is not a perfect system, but no regulatory system is perfect.
In fact, if you look at how the other regulatory systems for devices perform in the world, they perform no better than the CE marking system. The articles leave out a lot of things that would make them more balanced, like the fact that notified bodies are overseen and accredited by national authorities and that the authorities do diligently follow up every vigilance report that they receive.
A positive development of the Implant Files is more attention for regulation of medical devices, and specifically for resources committed by authorities. The CE marking system has so far performed very well with very little resources committed to it. This is a political choice that all of us are making together, see also what I wrote above here in this post. But it needs to be resourced properly for it to work. Look at the system for medicines: this is resourced much much better than the devices system and lo and behold, it’s the example for other systems. No shit Sherlock, you get what you pay for – also when it comes to regulatory systems. Also this is being addressed in the MDR and IVDR, by the way, with a renewed vigilance and market surveillance system. Now we just need some resources to roll out the system timely, as discussed above.
The Implant Files articles point at an increase of incidents in total, but they do not place it in the wider context of the increased use of implants for example, which is a missed opportunity to make it less sensationalist and more balanced. Oh wait – less sensation was not the point. Similarly, the assumption underlying the reporting seems to be that any incident in a treatment involving a device is attributable to the device only, while this if of course not necessarily true. Doctors may choose a perfect device for the wrong reasons, as a result of which the treatment will not deliver the results aimed for and the device is blamed. Or, what I often see in my practice: the device may fail as a result of the doctor’s fault. These cases are reported as incidents but are not attributable to the device, even if the device is a causal link in the damage.
It would also have been interesting if they would have compared medicines side effects to medical devices side effects so you can actually say something about acceptability of level of side effects. Medicines, their side effects and medication errors kill loads of people every year. Yet, I don’t hear people advocate for autopsy on every person taking meds that dies outside the hospital, even though enormous amounts of people die from adverse reactions to prescription medicines. Yet, this was proposed in one of the Implant Files items for people with pacemakers / ICDs.
Medicines have more robust trails because they take forever and have lots of people in them, and they have arms, including a placebo arm! But the nuance is again lost there too: you have to test a medicine in a clinical trial delivering results based on statistics because we do not fundamentally fully comprehend human biochemistry and make it transparent, so it’s actually a very crude mechanism. It’s like throwing someone out of an airplane to test a parachute. Maybe a lot of people, so it’s statistically sound. And then we throw some people out without parachute but tell them that we’re not sure if they have one or not to correct for the placebo effect. That is – conceptually – how we test medicines for a lack of better methods.
And did you know by the way that regardless of all that rigorous testing medicines remain incredibly dangerous and kill loads of people? The European Commission estimates that adverse reactions from prescription drugs cause 200,000 deaths per year in the EU, making prescription medicines drugs a major health risk, ranking 4th with stroke as a leading cause of death. Maybe not the best product to compare medical devices too, because I bet medical devices are not the 4th highest cause of death, not even by the estimation of the Implant Files. Would we like an approval mechanism that makes medical devices just as much of a universal killer as medicines? I bet we don’t. That is why these comparisons are so meaningless.
With devices however you can usually test if the device works or not because they work physically and not systemically, so you need fewer patients and less statistics. It would be quite immoral too to implant people with placebo pacemakers or placebo hips to see if the pacemaker or hip actually works. Because that is how we test medicines statistically for lack of a better way: you cannot observe biochemistry in action the way you can observe and test devices that act physically. It’s that simple, and that’s why we do not need huge populations of trial subjects to arrive at statistically relevant conclusions about how biochemistry functioned and what effects it had. That is why it makes a lot of sense to test medical devices clinically against the standard of care to see if they improve care compared to available solutions and check for unpredicted side effects, but a trail to statistically figure out if they work in the first place is not that ethical mostly.
You can wonder whether some implantable devices based on what companies thought was well understood technology were followed up with patients long enough because they did lead to systemic issues – and we did. It’s why we are experimenting with increased post market clinical follow up requirements under the current rules already, are fixing this too in the MDR and are paying particular attention to new implants, which need more data in advance. The Implant Files are conveniently ignoring that the EU started a major program to improve quality of approval, oversight and clinical evidence for medical devices in 2012 and has been rolling that out ever since, culminating in the new Medical Devices Regulation that entered into force last year and will apply in all aspects soon. But a problem solved is of course no news, so that is why the efforts to improve legislation that has been going on for years is basically not mentioned in the Implant Files.
Another problem I have with the Implant Files is how the articles and the TV shows blend US and EU requirements as if they are one single thing and draw implicit conclusions from this weird blend for the EU state of affairs. For example, a US expert is interviewed about how you can misrepresent things on an FDA incident reporting form in the US by selectively ticking boxes concerning cause of death and it is implied that the same is possible and in fact happens in the EU. However, as a starting point the EU model form for incident reporting is structured very differently, and this is not even addressed, nor how you would use that form to manipulate your vigilance reporting. Yet, the message is that this happens in the EU as well. There is no evidence however in the reporting that I came across of how you would do this in the EU. This is strange because the FOI requests provided the journalists with a deluge of EU vigilance reports and forms, which they could have easily checked. But they didn’t. This is just an example of one of the many things that give a wrong impression and I think unduly so.
The above is just a first impression. I will write more about the Implant Files, if only to help correct the sensationalist picture created on often half baked understanding or representation of the EU regulatory system.



Thanks Erik, great summary of the state of the art!
A good summary of the implant files that echoed a lot of my thoughts. Very little context amongst the sensationalist headlines. As you correctly point out, funding for devices is minimal compared with medicines and therefore is not a fair comparison (for many reasons). Staffing of medicines:devices in the UK at the MHRA was 9:1, and had annual budget cuts from the Dept of Health forcing constant evaluation of how to operate whilst making everything better and safer.