![]() Many people ask me these days “Hey Erik, what is the status of the revision? When will this thing finally complete?”.
Many people ask me these days “Hey Erik, what is the status of the revision? When will this thing finally complete?”.
Well, that’s an interesting question I often ask myself too. Of course I have my crystal ball and some bits of information from here and there but that still allows me to speculate only.
So I decided to request access to the latest documentation on the Council negotiations on the revision on the basis of EU freedom of information legislation. The Council had just published references to a bunch of documents that seemed to reflect the latest state of play.
Stuck
I had already heard from sources close to the negotiations that they are pretty much stuck at the moment and the process is very slow going with many entrenched positions. Don’t forget, in 2014 the Council working group combed through each of the chapters of the medical devices regulation alone twice in about 20 meetings in total. That’s a lot of time and effort to not even get close to where you would like to be and achieve compromises that do not seem to happen.
All that seems to be completely confirmed by the rejection of my request for disclosure below. As you can see, even a rejection can provide a lot of information:


In other words, the process is so delicate at the moment that the member states do not want their positions in public. That leaves me with the impression that the process is currently like hedgehogs making love: very slow and very careful with a ‘great political will to make it work’ in the end.
The latest documents of the April meetings are not publicly accessible either. I could request those too but I already know the answer to that request I think.
I like the consolation though that the acts will be published when they are final – thank goodness for democracy and for deadpan bureaucratic statements.
So what are the member states stuck on?
Well, we had something of an idea by end of last year. In the presentations of the MHRA and BSI at the DIA Euromeeting in Paris beginning of April there was a good rundown of key outstanding issues:
- ingested products – should they automatically be in the highest risk class (as currently proposed)?
- ‘non-medical devices’ – what will be in Annex XV from the start; there may be a new category of so-called ‘common specifications’ (so non-technical common technical specifications) for these devices.
- companion diagnostics – still disagreement about scope of the definition – align with FDA or not, cover selection and monitoring?
- non-viable human tissues and cells – this will likely ‘be aligned with the ATMP regulation’, which itself is up for revision now.
- viable biologic substances – exclusion where these support the intended purpose of the product
- reprocessing of single use devices – member states like the Commission proposal (national prohibitions possible), but don’t agree about whether to make in-house reprocessing possible (I’m not going to those hospitals) and there is the European Parliament proposal to deal with (single use is like printing your own money, so everything is presumed reprocessible) to deal with. Remember, the trilogue still has to start and the Parliament will want to get something out of it too.
- genetic testing – Council position to take to trilogues but there seems to be a majority that feels that the EU has no business in national practice of medicine. On the other hand, Peter Liese, the IVD Regulation rapporteur feels very strongly about this amendment regardless of how unconstitutional it is, which he sees as his baby.
- implant card – provision of card vs provision of information
- Eudamed & UDI – member states still disagree about what link Eudamed should have to UDI and how much detail should be left to implementing acts (over which they have little influence)
- summary of safety and performance
- notified bodies – supervision of notified bodies is still subject of disagreement, and should notified bodies do clinical audits?
- pre-market approval – still one of the big ticket items, especially important to France: will we have scrutiny, scrutiny plus or a real EU devices agency like EMA?
- clinical investigations – how do we align the clinical trials regime with the Clinical Trials Regulation with its different concepts? What clinical evidence is acceptable and how do we deal with equivalence?
- post market surveillance – how prescriptive should the regime be and do we want regular (trend) reporting for high risk devices?
- market surveillance & vigilance – how do we get to a coherent regime and how do we deal with risk vs incident?
- reference laboratories – this is still being heavily debated: should there be a split between reference and testing labs? Do we even need reference labs (mainly an IVD item) for medical devices?
- hazardous chemicals – big item for industry, member states divided over options of ban, phase out or labeling
- classification rules – what devices get up classified (looks like implants, surgical instruments)?
- governance and oversight – where do we get the money and the people to staff whatever it will turn out to be in the end?
This is a pretty long list of rather fundamental things to disagree about if you ask me. That is why I’m not super optimistic about a quick solution after the heroic crash and burn attempts of the Greek and Italian presidencies and the methodic current Latvian salami approach to the dossier, but hey, you never know. And it’s good to see that there is also at least some debate about the IVD Regulation. The MHRA speaker at the DIA Euromeeting compared the interplay between these issues to a game of Whack a Mole – when you think you’ve dealt with one, another pops up (again).
At least it seems there is no disagreement about the craziness of enormous scope extension of the definition of medical device proposed by the Parliament – I hope.
Timing
Time is of the essence by now – and this is the best time table we have for the moment (MHRA prediction from the DIA Euromeeting conference):
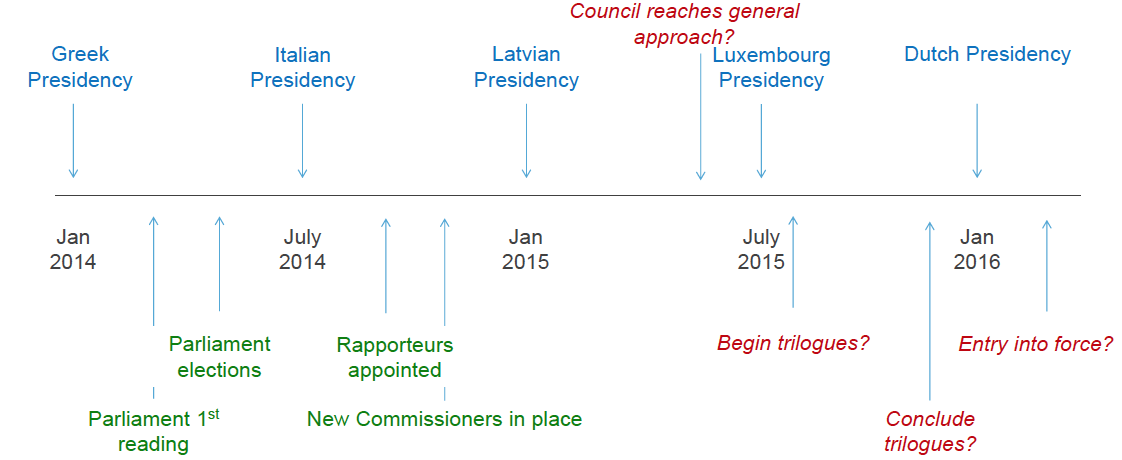
But will it blend?
But will it blend into compromises, these entrenched positions in the Council? And even then, what will the trilogue with the Commission and Parliament look like? You’ll remember that the Parliament deliberately took a completely different and far out approach on important points like hazardous substances, pre-market approval, notified bodies and genetic testing, just to mention a few. Difficult to see what the compromises on those points will look like, also because the rapporteur for the medical devices regulation dossier has changed in the mean time. I think I’ll have to boot up the crystal ball again for some more theorizing about where the institutional actors’ great political will to make it work will bring us – and when.



One comment
Do you want to comment?
Comments RSS and TrackBack URI
Trackbacks