
It had been been in the works for some time, although it also seemed unlikely for quite some time that this would happen. The implementation of the IVDR had been the slow little, neglected sister of the MDR implementation with greatly insufficient notified body capacity becoming available, and crucial elements of regulatory infrastructure (like the reference labs) still missing.
The MDCG published the Joint Implementation Plan for the IVDR in which it stated that there were serious issues with the IVDR that needed resolution.
Then the Council requested the Commission to prepare a legislative solution to deal with the problems plaguing the IVDR at the EPSCO Council of 15 June 2021. And also the European Parliament asked for a solution.
After an assessment of data on market readiness collected by the European Commission during the first half of 2021 the Commission concluded that Member States, health institutions, notified bodies and economic operators will not be in a position to ensure the proper implementation and application of the Regulation from 26 May 2022.
Now the proposal has been made public.
Let’s take a closer look at the proposal and see what we can say at this stage:
Is the proposal the anticipated “delay”?
Many have been hoping for a ‘delay’, often without being very clear of what they hoped that the delay would look like. The proposal is not a delay (because the date of application of the IVDR does not change – remains 26 May 2022), but something much more sophisticated and certainly not a one size fits all solution. The proposal does not simply move the date of application with one year as happened with the MDR (although there were additional tweaks too with the MDR). Given:
“the limited notified body capacity, the number of devices that need to undergo a conformity assessment involving a notified body should be spread over a longer period, allowing for a gradual phase-in of the new Regulation’s requirements while prioritising high-risk in vitro diagnostics. This can be achieved by amending Article 110 of the Regulation on transitional provisions, providing a period for existing higher risk class devices that is shorter than the one for existing lower risk class devices. At the same time, the existing transitional period for devices covered by notified body certificates issued under Directive 98/79/EC should be extended by 1 year until 26 May 2025. This will avoid that the transitional periods under Regulation (EU) 2017/745 and Regulation (EU) 2017/746 end at the same time and lessen the strain on Member States’ competent authorities, notified bodies, manufacturers, health institutions and other actors who deal with both medical devices and in vitro diagnostics.”
Proposal, p.4
Accordingly, the proposal is a combination of measures:
- IVDR risk class based phase-in (much like happened under the December 2019 MDR corrigendum for up-classified class I devices); and
- moving the backstop date of the IVDR grace period (to “lessen the strain on Member States’ competent authorities, notified bodies, manufacturers, health institutions and other actors who deal with both medical devices and in vitro diagnostics “, Proposal, p. 4).
These changes are made in article 110 IVDR, the article that sets out the transitional regime. If you apply track changes to the text of article 110 (2) – (4) IVDR as the proposal changes them this looks as follows:

No provisions seem to be made for ‘normal’ class A devices (only for sterile ones), which means that the date of application is set in stone at 26 May 2022 for normal class A devices, for example the lab instruments. Special categories of devices, such as companion diagnostics, seem to be addressed in the proposal by their risk class (which would be C for companion diagnostics).
As said and as you will have seen, the proposal is not a one size fits all solution. This means that every IVD company (manufacturers, but also importers and distributors (see below under legacy devices) )will need to check for each of its IVDs in which class it will end up in order to be able to see what the consequences of the proposal are (provided that the proposal is not altered in the legislative procedure of course). This means that you get to work with the MDCG IVDR classification guidance, which has been described in a lot of detail in the annotation of Annex VIII of the IVDR in my book The Enriched MDR and IVDR.
Legacy devices under IVDR
The result of the proposal is that unlike as originally intended under the IVDR there will now be a pretty large group of legacy devices on the market that will need to apply certain parts of the IVDR already as per date of application. The parts of the IVDR that have to applied for legacy devices will be bigger than is evident at sight, because an MDCG task force has issued an internal document in September stating that legacy devices under the MDR are subject to for example most parts of article 13 (importers) and 14 (distributors) MDR as well, which means that the same will apply for the IVDR. Unfortunately this document is not public (yet). It will however serve as input for other MDCG guidance, such as (I would expect) the impending MDCG guidance Q&A on importers and distributors.
While the document does not explicitly address the IVDR it would not be logical if it would not apply to the IVDR in the same way, since the economic operator provisions are identical and supposed to be interpreted the same. So, if you were betting on your legacy devices being out of scope of economic operator provisions of the MDR and IVDR, think again.
As I have seen with the MDR, manufacturers of legacy devices have found it very difficult to apply the article 120 (3) mandated parts of the MDR QMS that amended article 110 (3) IVDR would impose:
“the requirements of this Regulation relating to post-market surveillance, market surveillance, vigilance, registration of economic operators and of devices shall apply to devices referred to in the first, second and third subparagraph instead of the corresponding requirements in Directive 98/79/EC”.
Article 110 (3) IVDR
In addition, the legacy devices cannot undergo any significant changes until thy are IVDR CE marked (see for the MDCG guidance for the MDR on significant changes (not yet ported to the IVDR – MDCG?). This means a lot more than most people think. For example, you will not be able to do important software upgrades to a software IVD device as this will almost always trigger a significant change. If you implement your merger or acquisition wrong, you may well trigger a significant change that may invalidate the commercial rationale for the merger or acquisition. If you think you are buying an IVD company with legacy devices and the seller advertises that the company will implement important upgrades to their legacy devices in the coming years, this will likely not be possible due to the significant change restrictions, so buyer beware!
In-house produced devices
The Commission proposes more time to transition for the in-house produced devices regime under article 5 (5) IVDR:
“As, since its outbreak, many health institutions, in particular hospitals, have had to focus all their efforts on dealing with COVID-19, the Commission proposes to also introduce a transitional period for the requirements for devices manufactured and used within the same health institution (‘in-house devices’). This will give health institutions extra time to comply with the new requirements and ensure that in-house tests, which are often essential –especially for rare diseases, can continue to be developed in clinical laboratories.”
Proposal, p. 3
This extra time is implemented by changing Article 113(3) IVDR, the article that manages what provisions are delayed applicable, by adding points (i) and (j): ‘
- Article 5(5), points (b), (c) and (e) to (i), shall apply from 26 May 2024;
- Article 5(5), point (d), shall apply from 26 May 2028.’.
In non-lawyers language this means that most of the points of article 5 (5) are delayed until 26 May 2024 and one until 2028, but not all of them! Article 5 (5) (a), the introductory paragraph of Article 5 (5) (GSPRs in Annex I apply) and the final two paragraphs of Article 5 (5) will apply immediately as of the date of application of 26 May 2022.
This means that health institutions, while they do not have to have a suitable QMS ready for in-house devices by the date of application, the first restrictions on in-house devices are a fact by 26 May 2022: no more transferring to another legal entity, Annex I GSPRs documented, no industrial scale production and Member State controls on notification and type of in-house devices manufactured and used.
Preliminary discussion
Personally I still think it is surprising how long it took the MDCG, the Council and the Commission to arrive at the conclusion that the IVDR was in dire straits, heading to a situation of creating significant collateral damage in the transition. I have also marveled at how badly prepared health institutions have consistently been for the IVDR and how hard it has been for them to come to grips with being directly regulated under the MDR and the IVDR.
The proposal will need to take its time now to go through the legislative procedure, which is normally a process of at least a year. However, as we have seen with he MDR delay proposal last year, this can go a lot faster with the right amount of political alignment having taken place beforehand (two and a half months give or take). The question is of course if the political alignment is there. While the European Parliament has asked for a ‘delay’ this may not be exactly what they had been looking for. Also, the European Parliament had felt kind of ambushed with the MDR delay situation, and expressed a wish to have more time the next time around (which is this time). This defines the window as anything between two and a half months from now and the date of application of the IVDR.
Will this proposal, if it makes it into law, save the IVDR’s transition?
It will, first of all, depend on critical infrastructure being delivered on time. Eudamed is still a total tricky headache, even if modest steps are made and more modules are available on a voluntary basis now (devices and certificates come online beginning of October). The current experience is with the MDR, but the IVDR is still somewhat different so lots of new issues to discover. It would help if sufficient reference lab capacity is available timely. There is still a lack of guidance on the IVDR, although a lot of the MDR guidance can be leveraged to understand similar mechanisms in the IVDR (I have made convenient comparison tables for this in my book).
Secondly, a necessary condition is that the IVDR does not fall in the same trap as the MDR with a huge pile-up of legacy devices at the end of the transitional period that has a good chance of not making it to a regulation certificate because of lack of capacity, see the Team-NB position paper on expiring certificates, which shows the huge bulge at the end of the grace period:
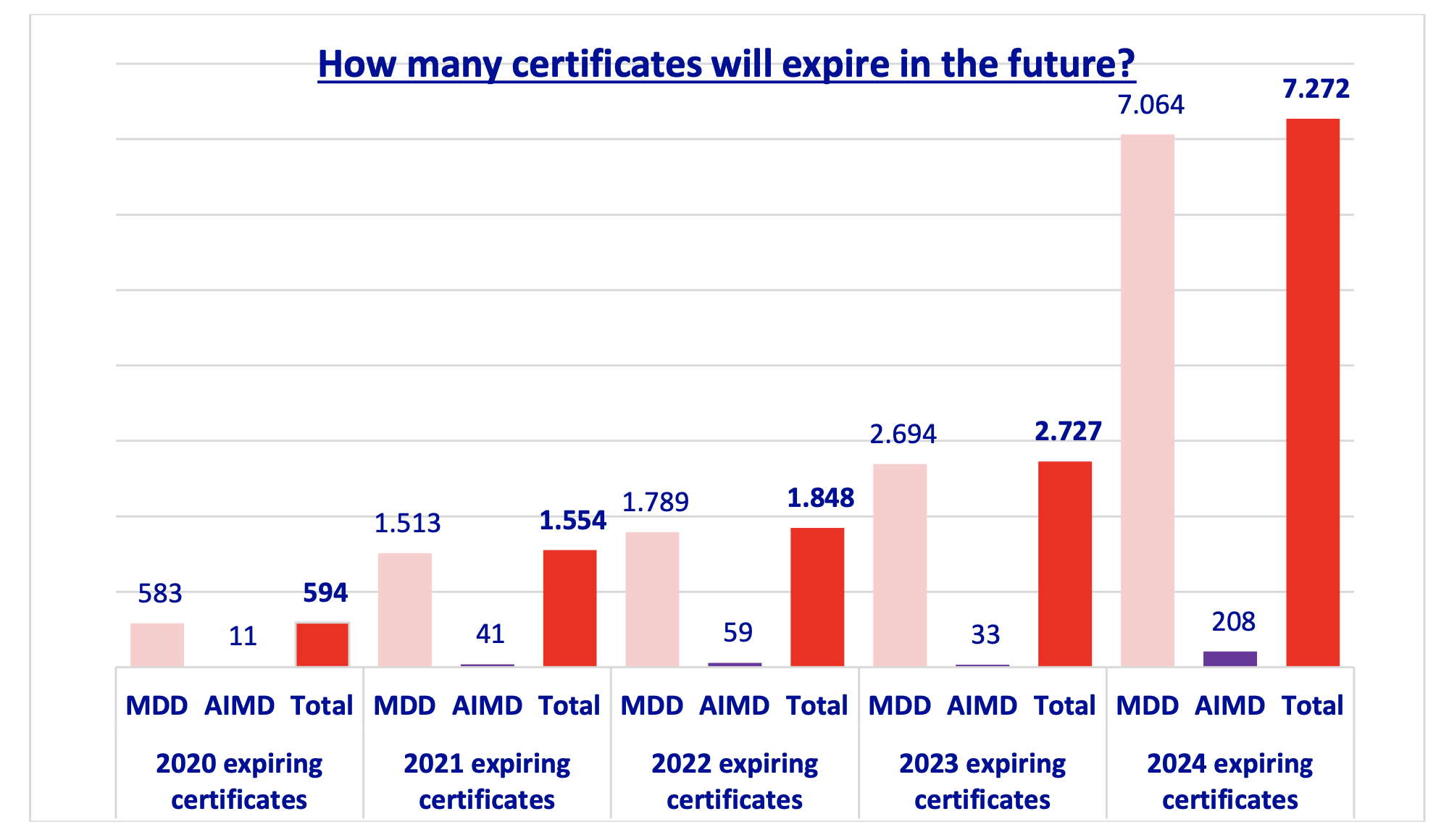
Directives expiring certificates (Team-NB)
The staggered phase-in of IVDs by class will hopefully make somewhat of a difference for the IVDR, but in the end it depends on when the notified body capacity is available. As we have seen with the MDR, you cannot kick the can down the road and then bet on sufficient capacity being available later – which is precisely what happened for the MDR. And now the question is whether the system can pass this huge stone without too much collateral damage.
Thirdly, more notified bodies capacity is needed than there currently is. There is nothing that this proposal does to improve this. This capacity will not only need to be able to deal with the enormous amount of manufacturers that has never dealt with a notified body before (and will make every time consuming mistake in the book) but also with new manufacturer coming to the market with new products. I personally see in my practice for the MDR and IVDR at this moment that new manufacturers (especially the small and medium sized ones) needing a notified body basically have very little chance of getting on board at a notified body, impacting their time to market.
So, early days to say if this proposal will save the day. It will really depend if the MDCG, the Commission and the Member States will put the necessary resources on the table to make the transition a success and whether industry and health institution will also commit the resources to understand and apply the rules timely. Rules without resources are basically wishful thinking.
Can you drop preparation and sit on your hands now as manufacturer? Better not, this is a proposal – it may not make it. If not, you will be faced with the current implementation schedule. So continue on your timelines for the moment. If you are an importer or distributor of legacy IVD devices, start learning because you will have a role to play in the system as well.



Thanks Erik! Thanks goodness sense prevailed
Hi Erik,
Thanks for getting this abstract and gap analysis out on such a short term! The IVDR proposal does provide the medical community with some air to breathe, but do not get your hopes up too high, the devil is always in the details. It mostly addresses the transitional provisions and gives some relief to LDT’s. I can imagine manufacturers, still, want to push forward to get their new IVD devices placed on the EU market, through self-declaration, before May next year…
Hi Erik â Thanks much as always for these great reads! Just a note on your comment to LTDs, I understand that 5(5)(d) is delayed until 2028 â only (a) applies from 2022, â but you seem to suggest that it applies already in 2022? Am I missing something? Best regards, Josefine
JOSEFINE SOMMER
Senior Associate
(Member of New York Bar, Brussels B List)
SIDLEY AUSTIN LLP
+32 2 504 6427
josefine.sommer@sidley.com
Hi Josefine, you are right, I missed that in the first version of this blog and then corrected it.