
We live in interesting times for medical devices in the Union – some have said we are headed for a cliff edge at the end of the grace period. I would personally prefer to say that we are in the danger zone, which would be a zone of danger (Archer pun on Top Gun), but still with some options. If you still need to see the new Top Gun movie: there are spoilers ahead, but only in the conclusion.
But I digress already – back to the story of this blog.
Not so long ago, in a galaxy not so far away before the date of application, everybody and their mother enthusiastically went all in on what we later understood to be legacy devices and (AI)MDD certificates were extended well into (or often all the way for) the ‘grace period’ provided under Article 120 (3) MDR. My analysis with hindsight knowledge is that many went into this in good faith, hoping that the issues plaguing the MDR as unfinished symphony with insufficient notified bdoy capacity would have been fixed by the time the legacy device certificates would expire. Others will have been happy to kick the can down the road and see where things are at by that time. Authorities and notified bodies will probably have been happy with the additional time to finalise the system.
And now here we are with a system that has still not been finalised and is not at capacity to pass the bulge of legacy certificates that need to be re-issued under the MDR, many of which will expire at the last possible date under the grace period. Already in 2020 the MDCG had attention for this problem in the Joint Implementation Plan for the MDR, but was then still in the groove that there was not enough data.
This became increasingly supported by evidence in several Team NB publications, with the latest one painting a dire picture that, regardless of notified body efforts to scale as much as they could, not even (roughly speaking) half of the total capacity required for 2024 would be available:
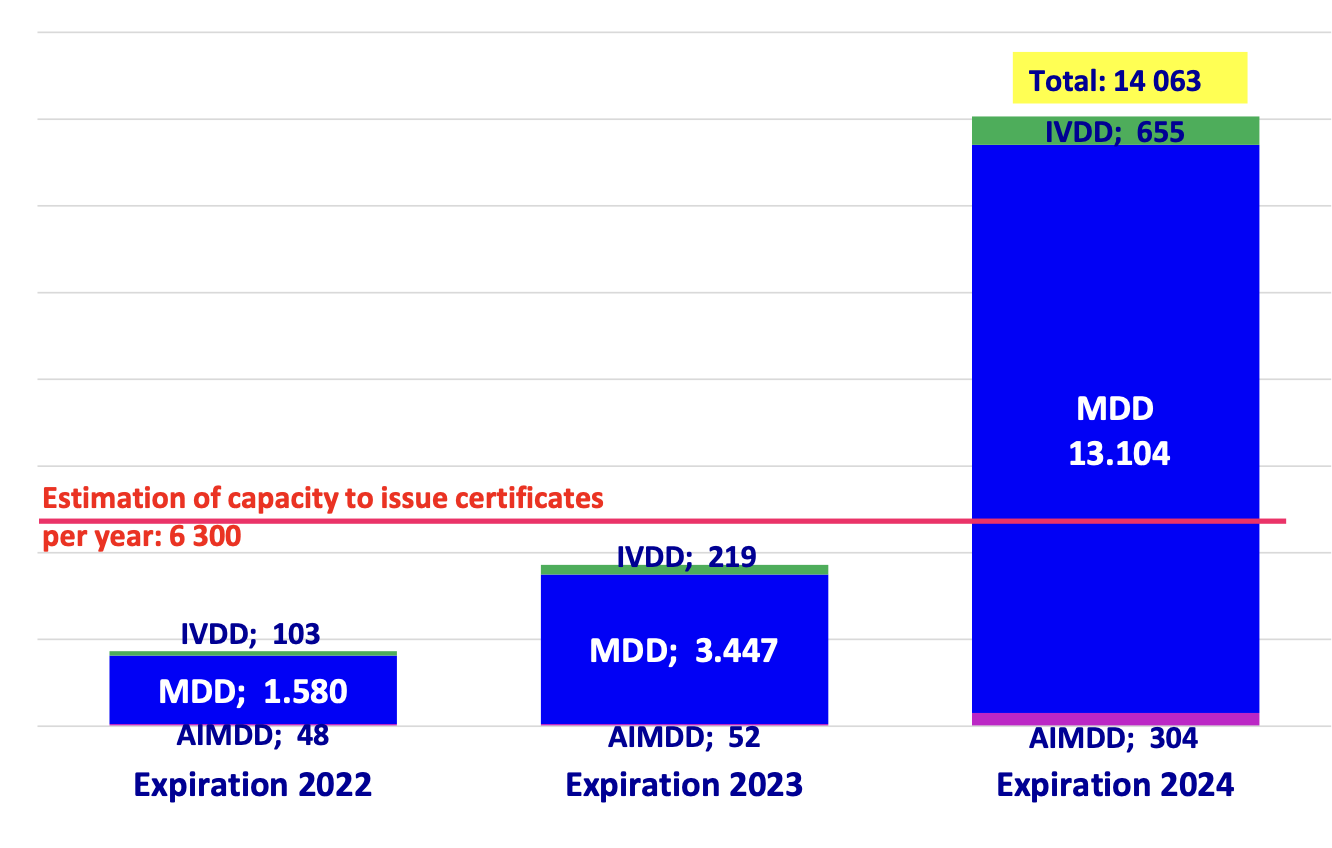
The authorities reacted first through a relatively mellow CAMD statement on the occasion of the CAMD’s 50th meeting (let’s all work together to fix this but the MDCG should take the lead), but then followed this up via the MDCG (which includes the voice of the Commission that presides the MDCG) with position paper MDCG 2022-11 that has a very different tone.
‘Sweeping your own little street clean’
The MDCG 2022-11 position paper document that was recently issued is certainly something else. There is some serious fingerpointing going on the document (it was not called ‘Notice to manufacturers to ensure timely compliance with MDR requirements’ for nothing) with the MDCG suggesting that many manufacturers are applying to notified bodies too late and/or with substandard quality submissions:
“The transition period intends to give further time to the system to prepare and to get ready, for example time for manufacturers to prepare their quality management system (QMS) and technical documentation before applying to a notified body. This step should not be perceived as a “grace period” to postpone the entering into application of the new rules. At this stage, data collected by notified bodies, and presented to competent authorities in December 2021, shows that nearly 37% of manufacturers’ applications have been refused on the basis of incomplete applications, underlining an overall lack of manufacturers’ preparedness. In April 2022, 75% of notified bodies indicated that more than 50% of the submitted applications were deemed incomplete.”
MDCG 2022-11, p. 2
In other words: you industry, stop sitting on yours hands and cease misunderstanding requirements! Grace periods are not for idling but for sorting out your problems so you don’t make them ours too. You can do better than this and have yourself to blame as much as you’re blaming others.
The paper is a nice exercise in political plausible deniability, or as we would say in Dutch in literal translation: ‘sweeping your own little street clean’.
Of course this does not mean that the MDCG is not right in saying that everyone is in this together, and that hurrying up and taking the transition to the MDR seriously is a very good idea under all circumstances. I personally still see too many companies where the sense of urgency has not really caught on yet, especially not with management. But then again, I also see a regulatory system that is excellent in its underlying concepts but implemented rather sub-optimally, which also causes all kinds of issues that the MDCG could have done a much better job at solving.
Some seeming light at the end of some tunnel
However, it’s not all bad news : MDCG 2022-11 gives us an insight in how the MDCG (which includes the Commission) thinks to solve the issues, which was not clear in the latest CAMD document or any other document before that.
The MDCG position paper implies that anyone that was hoping for a blanket extension of the grace period can stop hoping now: it’s not going to happen. I’ve been informing clients in this vein for some time now but now you can take some else’s word for it too.
Also, national and European exemptions under Article 59 MDR and national equivalents always have been an option, but only to support basic healthcare in Member States, not as an exemption for a regulatory pathway. It exceptional character was already clear in the guidance on Article 59, for example MDCG 2020-9, and this is now spelled out very clearly by the MDCG:
“Derogation from the conformity assessment procedure in accordance with Article 59 of the MDR has been mentioned as a possible remedy in case transition from AIMDD/MDD to MDR is not completed in time. It is important to stress that derogations may be granted by competent authorities only if the use of the device concerned is in the interest of public health, patient safety or patient health. This mechanism should not be considered as a solution for cases of late application to a notified body for conformity assessment or delays in the conformity assessment procedure. Economic grounds alone cannot justify a derogation under Article 59 MDR either.”
MDCG 2022-11, p. 3
But there seems to be something else in the works: the MDCG 2022-11 documents pretty explicitly hints at an implementing act under Article 97 (3) MDR in the works, putting the new market surveillance provisions and their harmonisation options in the MDR to good use:
“Also other mechanisms provided by the MDR in chapter VII (e.g. to deal with formal non-compliant products) will only be applicable for devices for which the manufacturer can demonstrate that he has undertaken all reasonable efforts to successfully conclude the transition to the MDR, including update of its QMS, in time. In this context, it is expected that the manufacturer has submitted an application to a notified body for certification in compliance with the MDR at least one year before the expiry date of the MDD/AIMDD certificate.”
MDCG 2022-11, p. 3
The reference to formal non-compliant products is a clear reference to the heading of Article 97 MDR, which provides in section 1:
“that a device does not comply with the requirements laid down in this Regulation but does not present an unacceptable risk to the health or safety of patients, users or other persons, or to other aspects of the protection of public health, they shall require the relevant economic operator to bring the non-compliance concerned to an end within a reasonable period that is clearly defined and communicated to the economic operator and that is proportionate to the non-compliance.”
Article 97 (1) MDR
Section 3 of Article 97 MDR provides the legal basis for the implementing act that seems to be under consideration:
“In order to ensure the uniform application of this Article, the Commission may, by means of implementing acts, specify appropriate measures to be taken by competent authorities to address given types of non-compliance. Those implementing acts shall be adopted in accordance with the examination procedure referred to in Article 114(3).”
Article 97 (3) MDR
An implementing act is a streamlined way to implement non-essential elements of EU regulation, while still giving the Parliament and the Council a degree of control over the Commission proposal. This is especially the case of implementing acts under Article 97 (3), which are subject to the heaviest comitology procedure, the examination procedure. Implementing act procedures are described in my book in a fair amount of detail – I recommend looking at the commentary to Article 114 MDR if you want to know more.
Unclear aspects remaining
However, some aspects in the outline given in MDCG 2022-11 still remain unclear.
For one, it is unclear when an application ‘has been submitted to a notified body’, especially given the large number of applications deemed incomplete as referenced by the MDCG. An implementing act should solve the issue with respect to the quality of the application and perhaps the manufacturer’s track record. If the implementing act merely requires an application ‘in the door’ at least one year before 26 May 2024 I can predict that a lot of not very high quality applications will be received at notified bodies on 25 May 2023. Applications eligible for exemption should probably have been validated somehow by the notified body or the competent authority, like you would normally see with marketing authorisation applications for medicinal products. Also, since the competent authorities would effectively be orphaning devices after expiry of the (AI)MDD certificate, I would expect that they would only do this for devices with an exemplary vigilance and safety record. How else would you meet the Article 97 (1) MDR requirement that the device does not present an unacceptable risk? Someone needs to make that call. That means that we can also expect an implementing act to contain requirements similar to those for orphaning of devices when the notified body lays down service or collapses uncontrolledly. Maybe the implementing act exemption would be combined with an orphaning-like application at national level, who knows.
Also, I hear more and more that notified bodies delay the either signing of the certification agreement or of validation of the application, in order to surprise manufacturers that think that they are on board with a message that the notified body is not going to process their application after all. If you look at the MDR closely (Annex VII 4.3 1st and 2nd para) on this subject, it is not clear how the certification agreement and the ‘formal’ application relate to each other. It’s clear that you cannot have one without the other, but the chicken and egg problem (which comes first) is not resolved clearly in the MDR. Any implementing act planned should resolve this. This is also especially relevant because Annex IX 2.1 requires the manufacturer to commit to the notified body exclusively when the application is made, leaving the manufacturer without alternatives.
Another problem is that the implementing act should not leave manufacturers out to dry when they submit a timely good faith application, in which the notified body still finds a minor flaw and tosses out the application for its own load management purposes at a moment where timely application to another notified body is not possible anymore because all will be occupied. This is connected to another problem: where will the notified bodies get the capacity to quickly validate all these new applications that will inevitably find their way to them early 2023 and how to avoid that this leads to crowding out of new applications for new devices even more than is already the case? Already now I see an increase in notified bodies telling manufacturers that they will just not have the capacity to process their MDR application.
Yet another unclear aspect is for how long notified bodies and manufacturers could expect any exemption under an implementing under Article 97 (3) to last, because Article 97 (1) MDR requires that the exemption is limited in time. Theoretically this could be ‘for as long as the notified body takes to finish the process’, but perhaps the MDCG would like a qualified backstop date in there not to draw out the process indefinitely.
Finally, hair-splitting lawyers with a passion for constitutionally correct legislative procedure like myself could wonder if Article 97 (3) MDR indeed has a constitutionally sufficiently strong basis to put in place an emergency valve in the transitional regime for the MDR. I personally am not sure if the delegation set out in Article 97 (3) MDR indeed was intended to include this type of measures but was aimed at a more modest harmonization of national market surveillance policies. On the other hand we’ve seen crazier actions from a constitutional perspective than this to fix MDR transition problems, like the bail-out of the class I up-classified classifieds by means of a corrigendum in December 2019. If there’s one thing that the history of the MDR has shown is that when push comes to shove, everybody will play along and is willing to get very creative.
Consultation during the summer?
By the standards of the Better Regulation Toolbox initiative that the Commission has imposed on itself, also implementing acts are subject to consultation (see Chapter 7, p. 439). So please Commission, make sure to engage in consultation and don’t do it in the summer holidays but preferably before.
I’m pretty sure some things have been overlooked that could benefit from consultation. Skipping consultation altogether would be not very nice by the Commission’s own standards.
But what about new, innovative devices?
Will the expected draft implementing act or other solution do anything for new devices with new applications, in other words: the non-legacy devices? What we are currently seeing in real life is that a growing number of companies, especially SMEs, is not getting on board with notified bodies because they are refused for lack of capacity. In other words, is MDCG 2022-11 announcing that a solution will only be available to manufacturers with legacy devices?
The Dutch competent authority IGJ seemed to suggest in a recent LinkedIn post alerting the market about MDCG 2022-11 that manufacturers with other than legacy devices will not be part of the solution hinted at in MDCG 2022-11, while MDGC 2022-11 clearly addresses all manufacturers:
“it is essential that all manufacturers adjust their system, finalise transition to the MDR and apply to a notified body, submitting complete and compliant applications, as soon as possible and well in advance of the end of the transition period to ensure timely compliance with the MDR”
MDCG 2022-11, p. 3
This does not necessarily mean that there might not be a solution for them in the works as well, but it would be nice to have clarity on that, right?
If there would not be a solution in the works for these devices, this would be extremely unfair in my opinon. Not only manufacturers with legacy devices currently have problems with notified bodies handling their applications timely. In fact many companies, often small and medium-sized enterprises, do not get on board at all with notified bodies at the moment and would be disadvantaged compared to manufacturers with legacy devices, for which the applications under the implementing act would contribute to the crowding out of applications for new devices. More often than with legacy devices, these are innovative devices that can add a lot of value for patients – it’s called innovation.
I have immediately asked the Dutch authorities how they see this but silence on that end so far.
Time is of the essence
I understand from reliable sources that something is already in the works at the Commission, which is very good news. However, this also means that manufacturers that do not have a validated MDR application for a legacy device in the works at a notified body at this moment must understand that time is now truly of the essence. Count back with me:
- Application must be made (or have been validated, we don’t know yet) one year before 26 May 2024 so by 25 May 2023 OR before the expiry date of the legacy device certificate (which may be sooner – 25 May 2023 is the very last date possible but your certificate expiry date may be sooner);
- This means that in the best possible scenario you have until 25 May 2023 to have an application going (about 11 months from now) and in the worse case much shorter, depending on actual certificate expiry date. Can you have ship shape application ready in no time if you’ve been sitting on your hands playing chicken with the system until now? It’s going to be an exciting and challenging ride, I can tell you that much.
This means that if you still need to file for your MDR application, you are in the danger zone. Which is a zone of danger – and you might consider calling Kenny Loggins (more Archer puns totally intended). And what is your plan B and plan C if this fails? Scenario anyone?
Time is of the essence for the Commission too, because any implementing act will still take time to trickle through the comitology procedure and we’ll need something proposed more than ASAP if the industry and notified bodies are to be informed about their options in any way that is still somewhat timely and meaningful to do something with the options offered (see left limb in the below diagram for examination procedure):
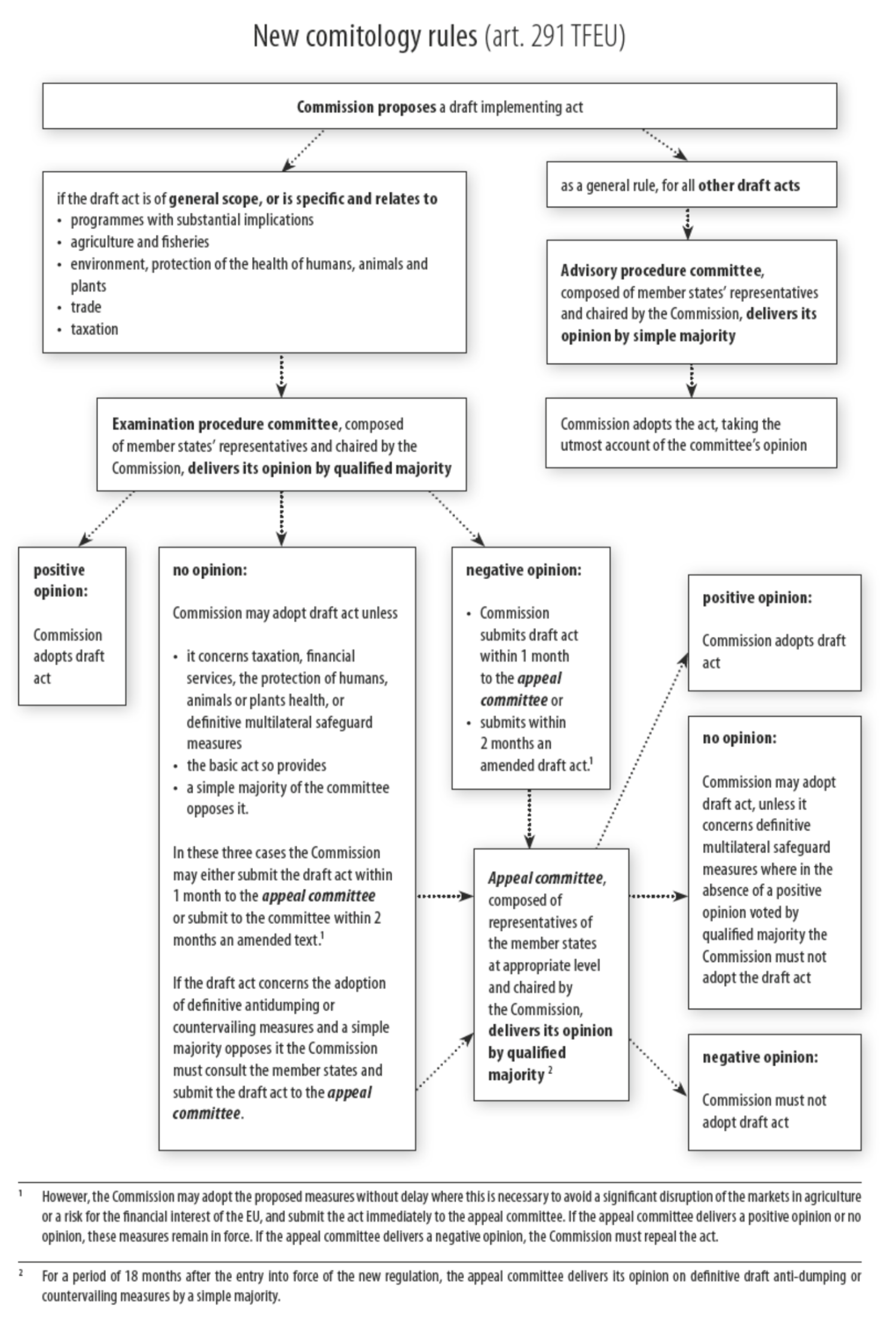
The need for speed
We have an interesting summer ahead in which all manufacturers, whether with legacy devices or not, are in the danger zone.
And as the saying goes in the new Top Gun movie (which prominently figures Kenny Loggins’ Danger Zone song): it’s not the plane, it’s the pilot.
So now it’s up to the brave pilots of industry regulatory departments to scramble whatever your company provides as operational jets (I know that not everyone can even hope for a latest version F-18 or better equivalent) and perform two miracles in a row like in that new Top Gun movie: first finalise your MDR applications in an impossibly tight time frame and then launch the applications into the notified body’s narrow application window in a veritable Death Star approach.
The same is true for the brave pilots at the Commission, who will need to scramble even faster to put the implementing act and/or whatever else is in the works according to MDCG 2022-11 together and shepherd it through the examination procedure because we will need to know where to go in order to be able to end up there.
And finally, there are the brave pilots at the notified bodies, who will be once again be pulling 9 G turns solving everybody else’s problems, having to see through the load of applications made in a transitional regime in which the goalposts keep being moved.
I hope everyone feels the need – the need for speed, because we’re is going to need it.



Nice interesting blog (once again:-); thank you for sharing your thoughts! Lately I hear a lot of people worrying about the date 26 May 2024. Offcourse I understand, but what I don’t understand is why nobody is mentioning article 120(4) MDR. I would think that after May 2024 it is no longer possible to place Medical Devices on the market, but Medical Devices that already have been placed on the market can still be put into service until May 2025. I understand this is not relevant for new Medical Devices, but for legacy devices this means that article 97 MDR could only be applicable after May 2025…or have I misinterpreted section 4 of article 120 MDR?
Thanks for another great post, Erik.
I’d like to comment on the issue of “low quality submissions”, as someone who’s been closely involved in the preparation/compilation, submission, initial “validation” by NBs, and finally the detailed NB review of “validated” submissions. While it’s true that sometimes QMSs and Technical Documentation (“Technical Files”) are not up to reasonable standards, all the way up to submission, NBs apparently have a lot of discretion/leeway as to what they pursue as “acceptable quality” of such documentation. My experience shows that in many instances, during both the initial “validation” (BSI refers to it as “completeness check”, and they have an explicit checklist for it and some detailed guidance) and the subsequent detailed review(s), the NB would exercise unreasonable nit-picking where the quality of the documentation is actually quite good, as far as MDR text (and intent, as far as we can tell) and MDCG guidance are concerned. The NB is far from powerless in this regard, and it’s not an impartial, “automatic” algorithm that’s being followed. Rather, it is more often quite a blurry process, where solid rationales (on the NB’s part) are “a luxury” manufacturers rarely get to enjoy, and the goalposts seem to be easily moveable.
Again, I agree that many times the manufacturer’s documentation is lacking. Sometimes it’s good-old procrastination or an attempt to save on resources where there’s really no way around it. But in many other cases it’s simply the result of the MDR raising the bar (by degrees?) relative to the MDD days, and please let’s not forget that 2020-21 (at least) have been tough on everyone, including team members who were supposed to generate all that added documentation and keep it well maintained. Many manufacturers are not yet out of the woods in that respect. Even with proactive, well-meaning and well-planning manufacturers, some gaps may persist, which leads to the the questions “Is all of this really necessary?” and “Is this really of essence?”. NBs seem to me to be only too happy to automatically reply yes and yes – why bother with nuances?… While the initial verification of a submission is a relatively straightforward step, this gets very murky very quickly once detailed review begins, and while the infinitely-rigid approach is simple and risk-free from the NB perspective (no one will blame them for being partial or inconsistent), the certification timeline ramifications are disastrous. All in all, I think that a very big part of the problem is a matter of attitude. Perhaps the CAs are holding the NBs on an extremely tight leash – I wouldn’t know, because it’s not where I’m active; but the end result for the manufacturer it as described above. Perhaps a sea change is necessary on a Union level.
I’d also like to provide my personal perspective on the “lack of NB review capacity” issue, as built-up over the last 5 years. I hear that tune a lot. I find it very odd, though, that in spite of me sitting idle for months on end, and in spite of my availability having high visibility online (e.g. LinkedIn) and me being quite vocal about it online until 1-2 years ago (I stopped because it seemed pointless), not a single time have I been approached by anyone in any NB with a request to propose the provision of review services. I wonder why. Is it because I’m not an EU citizen? If that’s the answer, I see no sense in that. To be knowledgeable and experienced in implementing the MDR you don’t need any particular nationality. Further, the pandemic has demonstrated beautifully (to anyone who needed such a demonstration) that remote desktop work can be just as effective as in-person, in some cases, and is in most cases more efficient. I understand that in-depth Technical Documentation reviewing requires first a thorough vetting (theoretically; I’ve come across reviewers who obviously lacked some basic pre-requisites), but at least the stage of initial submission “validation” – which the post predicts is going to be a major bottleneck quite soon – can definitely be done effectively and efficiently remotely, by anyone with my kind of background. I even tried approaching NBs and offering my services proactively; however, you’d be able to count the number of replies I received on the fingers on your head (that’s right, it’s not a typo – your head). Not even polite “No, thank you”. So please forgive me that I’m not even slightly moved every time I hear about lack of NB review capacity. If one lacks capacity, and REALLY wants to do something about it, wouldn’t they leave no stone unturned in their search?…
Cheers,
Ronen.
Excellet Erik !
Excellent information. Thanks
Absolutely brilliant piece of analysis.
Sent from my iPhone
>